
ヒトの遺伝的変異
Human genetic variation
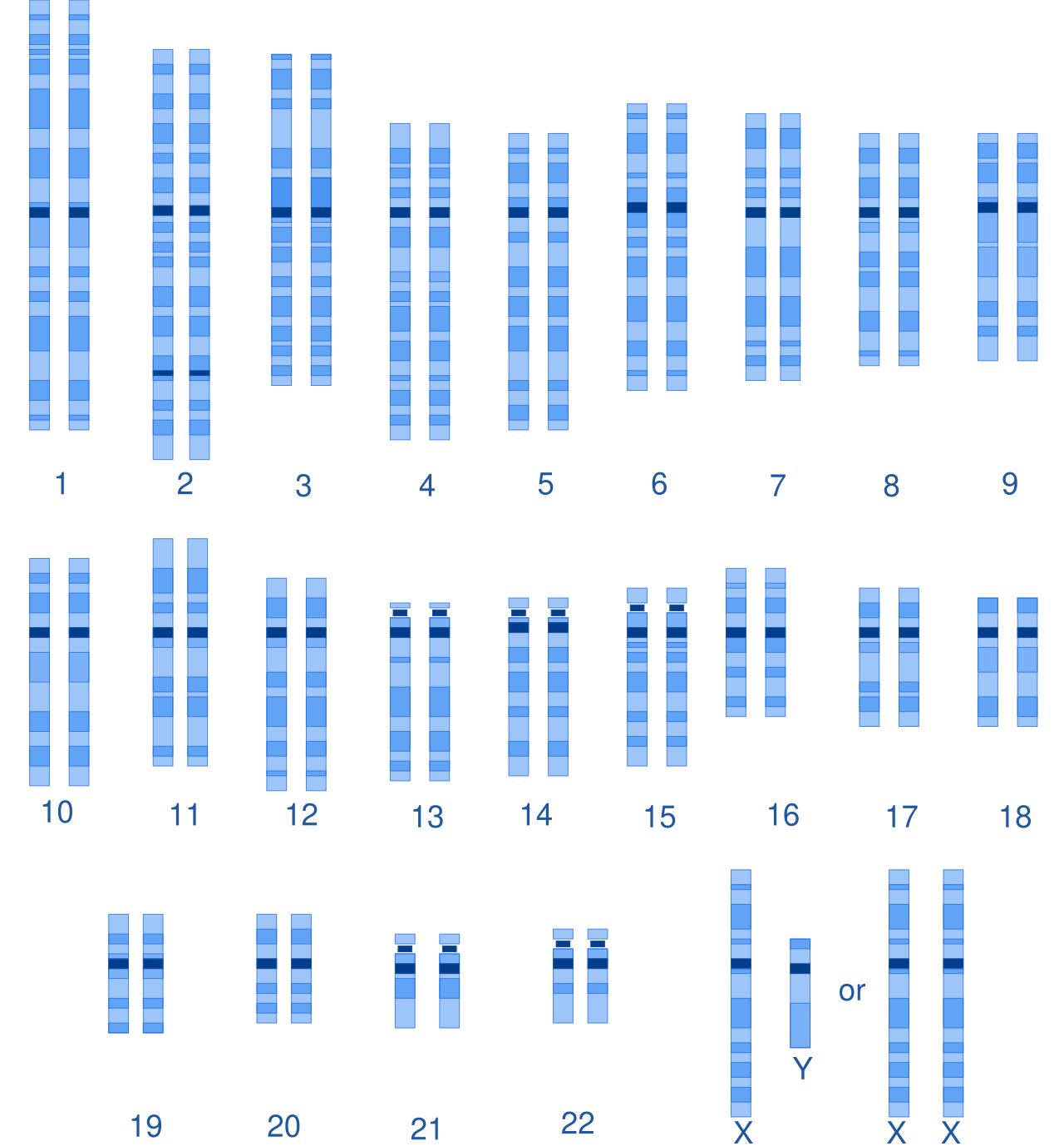
A graphical representation of the typical human karyotype/ The human mitochondrial DNA
☆ ヒトの遺伝的変異とは、集団内および集団間の遺伝的差異である。ヒトの集団には、ある遺伝子の変異体(対立遺伝子)が複数存在する場合があり、これは多型 と呼ばれる状況である。 遺伝的に同じ人間は2人といない。一卵性双生児(1つの接合体から発生する)であっても、発生過程で起こる突然変異や遺伝子のコピー数の変化により、遺伝 的な差異を持つことはまれである。 ヒトゲノムの全長は、46本の染色体DNAで約32億塩基対(bp)であり、細胞内のミトコンドリアには17,000bp弱のDNAが存在する。2015 年、個人のゲノムと参照ゲノムとの典型的な差異は、2,000万塩基対(または全体の0.6%)と推定された。2017年現在、配列決定されたヒトゲノム から、合計3億2,400万個の変異が知られている。 比較的、ヒトは遺伝的に均質な種である。少数の遺伝的変異は、特定の地理的地域やその地域の祖先を持つ人々により頻繁に見られるが、この変異がヒトゲノム の変異のごく一部(~15%)を占めるにすぎない。変異の大部分は、各ヒト集団のメンバー内に存在する。比較として、アカゲザルはヒトの2.5倍のDNA 配列の多様性を示す。チンパンジーは核DNAを解析した場合、ヒトよりも遺伝的多様性が高いが、ヒトはタンパク質レベルで解析した場合、ヒトよりも遺伝的 多様性が高い。 ヒト集団間の遺伝的距離に不連続性がないこと、ヒト種に不連続な分岐がないこと、ヒトが世界的に著しく均質であることは、ヒトに人種や亜種を推定する科学 的根拠がないことを意味しており、ほとんどの形質について、集団間よりも集団内の変異の方がはるかに大きい。にもかかわらず、現代の遺伝子研究では、皮膚 の色、体の大きさ、乳糖とデンプンの消化、高地適応、薬物反応、味覚受容体、特定の病気を発症しやすい体質などの形質において、ヒトの集団間でかなりの平 均的な遺伝的差異があることがわかっている。最大の多様性はアフリカの集団内および集団間で見られ、アフリカ大陸からの距離が遠くなるにつれて徐々に減少 していく。 ヒトの遺伝的変異の研究は、進化学的に重要であり、医学的応用も可能である。科学者が過去の人類の移動のパターンを復元し、理解するのに役立つ。医学の分 野では、ヒトの遺伝的変異の研究は重要である。というのも、病気を引き起こす対立遺伝子の中には、特定の集団に多く見られるものがあるからである。例え ば、鎌状赤血球貧血の突然変異は、サハラ以南のアフリカ、南ヨーロッパ、アラビア、インドの特定の集団に祖先を持つ人々に多く見られる。
| Human genetic
variation is the genetic differences in and among populations. There
may be multiple variants of any given gene in the human population
(alleles), a situation called polymorphism. No two humans are genetically identical. Even monozygotic twins (who develop from one zygote) have infrequent genetic differences due to mutations occurring during development and gene copy-number variation.[1] Differences between individuals, even closely related individuals, are the key to techniques such as genetic fingerprinting. The human genome has a total length of approximately 3.2 billion base pairs (bp) across 46 chromosomes of DNA as well as slightly under 17,000 bp DNA in cellular mitochondria. In 2015, the typical difference between an individual's genome and the reference genome was estimated at 20 million base pairs (or 0.6% of the total).[2] As of 2017, there were a total of 324 million known variants from sequenced human genomes.[3] Comparatively speaking, humans are a genetically homogeneous species. Although a small number of genetic variants are found more frequently in certain geographic regions or in people with ancestry from those regions, this variation accounts for a small portion (~15%) of human genome variability. The majority of variation exists within the members of each human population. For comparison, rhesus macaques exhibit 2.5-fold greater DNA sequence diversity compared to humans.[4] These rates differ depending on what macromolecules are being analyzed. Chimpanzees have more genetic variance than humans when examining nuclear DNA, but humans have more genetic variance when examining at the level of proteins.[5] The lack of discontinuities in genetic distances between human populations, absence of discrete branches in the human species, and striking homogeneity of human beings globally, imply that there is no scientific basis for inferring races or subspecies in humans, and for most traits, there is much more variation within populations than between them.[6][7][8][9][10][11][12][13] Despite this, modern genetic studies have found substantial average genetic differences across human populations in traits such as skin colour, bodily dimensions, lactose and starch digestion, high altitude adaptions, drug response, taste receptors, and predisposition to developing particular diseases.[14][12] The greatest diversity is found within and among populations in Africa,[15] and gradually declines with increasing distance from the African continent, consistent with the Out of Africa theory of human origins.[15] The study of human genetic variation has evolutionary significance and medical applications. It can help scientists reconstruct and understand patterns of past human migration. In medicine, study of human genetic variation may be important because some disease-causing alleles occur more often in certain population groups. For instance, the mutation for sickle-cell anemia is more often found in people with ancestry from certain sub-Saharan African, south European, Arabian, and Indian populations, due to the evolutionary pressure from mosquitos carrying malaria in these regions. New findings show that each human has on average 60 new mutations compared to their parents.[16][17] |
ヒトの遺伝的変異とは、集団内および集団間の遺伝的差異である。ヒトの
集団には、ある遺伝子の変異体(対立遺伝子)が複数存在する場合があり、これは多型と呼ばれる状況である。 遺伝的に同じ人間は2人といない。一卵性双生児(1つの接合体から発生する)であっても、発生過程で起こる突然変異や遺伝子のコピー数の変化により、遺伝 的な差異を持つことはまれである。 ヒトゲノムの全長は、46本の染色体DNAで約32億塩基対(bp)であり、細胞内のミトコンドリアには17,000bp弱のDNAが存在する。2015 年、個人のゲノムと参照ゲノムとの典型的な差異は、2,000万塩基対(または全体の0.6%)と推定された[2]。2017年現在、配列決定されたヒト ゲノムから、合計3億2,400万個の変異が知られている[3]。 比較的、ヒトは遺伝的に均質な種である。少数の遺伝的変異は、特定の地理的地域やその地域の祖先を持つ人々により頻繁に見られるが、この変異がヒトゲノム の変異のごく一部(~15%)を占めるにすぎない。変異の大部分は、各ヒト集団のメンバー内に存在する。比較として、アカゲザルはヒトの2.5倍のDNA 配列の多様性を示す[4]。チンパンジーは核DNAを解析した場合、ヒトよりも遺伝的多様性が高いが、ヒトはタンパク質レベルで解析した場合、ヒトよりも 遺伝的多様性が高い[5]。 ヒト集団間の遺伝的距離に不連続性がないこと、ヒト種に不連続な分岐がないこと、ヒトが世界的に著しく均質であることは、ヒトに人種や亜種を推定する科学 的根拠がないことを意味しており、ほとんどの形質について、集団間よりも集団内の変異の方がはるかに大きい。 [6][7][8][9][10][11][12][13]にもかかわらず、現代の遺伝子研究では、皮膚の色、体の大きさ、乳糖とデンプンの消化、高地適 応、薬物反応、味覚受容体、特定の病気を発症しやすい体質などの形質において、ヒトの集団間でかなりの平均的な遺伝的差異があることがわかっている。 [14][12]最大の多様性はアフリカの集団内および集団間で見られ[15]、アフリカ大陸からの距離が遠くなるにつれて徐々に減少していく。 ヒトの遺伝的変異の研究は、進化学的に重要であり、医学的応用も可能である。科学者が過去の人類の移動のパターンを復元し、理解するのに役立つ。医学の分 野では、ヒトの遺伝的変異の研究は重要である。というのも、病気を引き起こす対立遺伝子の中には、特定の集団に多く見られるものがあるからである。例え ば、鎌状赤血球貧血の突然変異は、サハラ以南のアフリカ、南ヨーロッパ、アラビア、インドの特定の集団に祖先を持つ人々に多く見られる。 新たな知見によると、ヒトは親と比較して平均60の新たな突然変異を持っている[16][17]。 |
| Causes of variation Further information: Recent human evolution Causes of differences between individuals include independent assortment, the exchange of genes (crossing over and recombination) during reproduction (through meiosis) and various mutational events. There are at least three reasons why genetic variation exists between populations. Natural selection may confer an adaptive advantage to individuals in a specific environment if an allele provides a competitive advantage. Alleles under selection are likely to occur only in those geographic regions where they confer an advantage. A second important process is genetic drift, which is the effect of random changes in the gene pool, under conditions where most mutations are neutral (that is, they do not appear to have any positive or negative selective effect on the organism). Finally, small migrant populations have statistical differences – called the founder effect – from the overall populations where they originated; when these migrants settle new areas, their descendant population typically differs from their population of origin: different genes predominate and it is less genetically diverse. In humans, the main cause is genetic drift.[18] Serial founder effects and past small population size (increasing the likelihood of genetic drift) may have had an important influence in neutral differences between populations. [citation needed] The second main cause of genetic variation is due to the high degree of neutrality of most mutations. A small, but significant number of genes appear to have undergone recent natural selection, and these selective pressures are sometimes specific to one region.[19][20] |
ばらつきの原因 さらに詳しい情報 最近の人類の進化 個体間の違いの原因には、独立した取り合わせ、(減数分裂による)生殖中の遺伝子の交換(交叉と組換え)、様々な突然変異現象などがある。 集団間に遺伝的変異が存在する理由は少なくとも3つある。ある対立遺伝子が競争上有利に働く場合、自然選択は特定の環境において個体に適応上の優位性を与 えるかもしれない。選択下にある対立遺伝子は、その対立遺伝子が優位性をもたらす地理的地域にのみ存在する可能性が高い。第二の重要な過程は遺伝的ドリフ トであり、ほとんどの突然変異が中立である(つまり、生物にプラスにもマイナスにも選択的影響を与えない)条件下で、遺伝子プールがランダムに変化する影 響である。最後に、小規模な移住者集団は、その起源となった集団全体との統計的な差異-創始者効果と呼ばれる-を持つ。これらの移住者が新しい地域に定住 すると、その子孫集団は通常、起源となった集団とは異なる:異なる遺伝子が優勢となり、遺伝的多様性が低くなる。 ヒトの場合、主な原因は遺伝的ドリフトである[18]。連続的な創始者効果と過去の小さな集団サイズ(遺伝的ドリフトの可能性を高める)が、集団間の中立 的な差異に重要な影響を及ぼしている可能性がある。[遺伝的変異の2番目の主な原因は、ほとんどの突然変異が高度に中立的であることである。少数ではある が、かなりの数の遺伝子が最近の自然淘汰を受けているようであり、これらの選択圧はある地域に特異的であることもある[19][20]。 |
| Measures of variation Genetic variation among humans occurs on many scales, from gross alterations in the human karyotype to single nucleotide changes.[21] Chromosome abnormalities are detected in 1 of 160 live human births. Apart from sex chromosome disorders, most cases of aneuploidy result in death of the developing fetus (miscarriage); the most common extra autosomal chromosomes among live births are 21, 18 and 13.[22] Nucleotide diversity is the average proportion of nucleotides that differ between two individuals. As of 2004, the human nucleotide diversity was estimated to be 0.1%[23] to 0.4% of base pairs.[24] In 2015, the 1000 Genomes Project, which sequenced one thousand individuals from 26 human populations, found that "a typical [individual] genome differs from the reference human genome at 4.1 million to 5.0 million sites … affecting 20 million bases of sequence"; the latter figure corresponds to 0.6% of total number of base pairs.[2] Nearly all (>99.9%) of these sites are small differences, either single nucleotide polymorphisms or brief insertions or deletions (indels) in the genetic sequence, but structural variations account for a greater number of base-pairs than the SNPs and indels.[2][25] As of 2017, the Single Nucleotide Polymorphism Database (dbSNP), which lists SNP and other variants, listed 324 million variants found in sequenced human genomes.[3] Single nucleotide polymorphisms  DNA molecule 1 differs from DNA molecule 2 at a single base-pair location (a C/T polymorphism). Main article: Single nucleotide polymorphism A single nucleotide polymorphism (SNP) is a difference in a single nucleotide between members of one species that occurs in at least 1% of the population. The 2,504 individuals characterized by the 1000 Genomes Project had 84.7 million SNPs among them.[2] SNPs are the most common type of sequence variation, estimated in 1998 to account for 90% of all sequence variants.[26] Other sequence variations are single base exchanges, deletions and insertions.[27] SNPs occur on average about every 100 to 300 bases[28] and so are the major source of heterogeneity. A functional, or non-synonymous, SNP is one that affects some factor such as gene splicing or messenger RNA, and so causes a phenotypic difference between members of the species. About 3% to 5% of human SNPs are functional (see International HapMap Project). Neutral, or synonymous SNPs are still useful as genetic markers in genome-wide association studies, because of their sheer number and the stable inheritance over generations.[26] A coding SNP is one that occurs inside a gene. There are 105 Human Reference SNPs that result in premature stop codons in 103 genes. This corresponds to 0.5% of coding SNPs. They occur due to segmental duplication in the genome. These SNPs result in loss of protein, yet all these SNP alleles are common and are not purified in negative selection.[29] Structural variation Main article: Structural variation Structural variation is the variation in structure of an organism's chromosome. Structural variations, such as copy-number variation and deletions, inversions, insertions and duplications, account for much more human genetic variation than single nucleotide diversity. This was concluded in 2007 from analysis of the diploid full sequences of the genomes of two humans: Craig Venter and James D. Watson. This added to the two haploid sequences which were amalgamations of sequences from many individuals, published by the Human Genome Project and Celera Genomics respectively.[30] According to the 1000 Genomes Project, a typical human has 2,100 to 2,500 structural variations, which include approximately 1,000 large deletions, 160 copy-number variants, 915 Alu insertions, 128 L1 insertions, 51 SVA insertions, 4 NUMTs, and 10 inversions.[2] Copy number variation Main article: Copy number variation A copy-number variation (CNV) is a difference in the genome due to deleting or duplicating large regions of DNA on some chromosome. It is estimated that 0.4% of the genomes of unrelated humans differ with respect to copy number. When copy number variation is included, human-to-human genetic variation is estimated to be at least 0.5% (99.5% similarity).[31][32][33][34] Copy number variations are inherited but can also arise during development.[35][36][37][38] A visual map with the regions with high genomic variation of the modern-human reference assembly relatively to a Neanderthal of 50k[39] has been built by Pratas et al.[40] Epigenetics Epigenetic variation is variation in the chemical tags that attach to DNA and affect how genes get read. The tags, "called epigenetic markings, act as switches that control how genes can be read."[41] At some alleles, the epigenetic state of the DNA, and associated phenotype, can be inherited across generations of individuals.[42] Genetic variability Main article: Genetic variability Genetic variability is a measure of the tendency of individual genotypes in a population to vary (become different) from one another. Variability is different from genetic diversity, which is the amount of variation seen in a particular population. The variability of a trait is how much that trait tends to vary in response to environmental and genetic influences. Clines Main article: Cline (biology) In biology, a cline is a continuum of species, populations, varieties, or forms of organisms that exhibit gradual phenotypic and/or genetic differences over a geographical area, typically as a result of environmental heterogeneity.[43][44][45] In the scientific study of human genetic variation, a gene cline can be rigorously defined and subjected to quantitative metrics. Haplogroups Main article: Haplogroup In the study of molecular evolution, a haplogroup is a group of similar haplotypes that share a common ancestor with a single nucleotide polymorphism (SNP) mutation. The study of haplogroups provides information about ancestral origins dating back thousands of years.[46] The most commonly studied human haplogroups are Y-chromosome (Y-DNA) haplogroups and mitochondrial DNA (mtDNA) haplogroups, both of which can be used to define genetic populations. Y-DNA is passed solely along the patrilineal line, from father to son, while mtDNA is passed down the matrilineal line, from mother to both daughter or son. The Y-DNA and mtDNA may change by chance mutation at each generation. Variable number tandem repeats Main article: Variable number tandem repeat A variable number tandem repeat (VNTR) is the variation of length of a tandem repeat. A tandem repeat is the adjacent repetition of a short nucleotide sequence. Tandem repeats exist on many chromosomes, and their length varies between individuals. Each variant acts as an inherited allele, so they are used for personal or parental identification. Their analysis is useful in genetics and biology research, forensics, and DNA fingerprinting. Short tandem repeats (about 5 base pairs) are called microsatellites, while longer ones are called minisatellites. |
変異の尺度 ヒトの遺伝的変異は、ヒトの核型の重大な変化から一塩基の変化に至るまで、様々な規模で生じている[21]。染色体異常は、ヒトの出生160例中1例で検出される。性染色体異常は別として、異数性の場合、発育中の胎児が死亡(流産)するケースがほとんどである。 ヌクレオチドの多様性とは、2つの個体間で異なるヌクレオチドの平均的な割合である。2004年の時点で、ヒトの塩基配列の多様性は塩基対の0.1% [23]から0.4%と推定されている[24]。2015年、26のヒト集団から1,000人の塩基配列を決定した1000人ゲノムプロジェクトは、「典 型的な(個人の)ゲノムは、参照ヒトゲノムと410万から500万箇所異なっている。 これらの部位のほぼすべて(99.9%以上)は、遺伝子配列における一塩基多型や短時間の挿入や欠失(indel)のような小さな違いであるが、SNPや indelよりも構造的な変異の方がより多くの塩基対を占めている[2][25]。 2017年現在、SNPやその他の変異体をリストアップしている一塩基多型データベース(dbSNP)は、配列決定されたヒトゲノムで見つかった3億2,400万個の変異体をリストアップしている[3]。 一塩基多型 DNA分子1はDNA分子2と1塩基対の位置が異なる(C/T多型)。 主な記事 一塩基多型 一塩基多型(SNP)とは、母集団の少なくとも1%に見られる、ある種のメンバー間の一塩基の違いのことである。1000ゲノムプロジェクトで解析された 2,504個体には、8,470万個のSNPが存在した[2]。SNPは最も一般的なタイプの配列変異であり、1998年には全配列変異の90%を占める と推定されている[26]。 機能的SNP、すなわち非同義SNPは、遺伝子スプライシングやメッセンジャーRNAのような何らかの因子に影響を与えるものであり、種のメンバー間で表 現型の違いを引き起こす。ヒトのSNPの約3%から5%が機能的である(International HapMap Projectを参照)。中立的なSNP、または同義的なSNPは、その数の多さと世代を超えて安定的に遺伝することから、ゲノムワイド関連研究における 遺伝マーカーとして依然として有用である[26]。 コーディングSNPとは、遺伝子の内部に存在するSNPである。103の遺伝子に早発停止コドンをもたらすヒトの参照SNPが105個存在する。これは コーディングSNPの0.5%に相当する。これらのSNPはゲノムの重複によって生じる。これらのSNPはタンパク質の損失をもたらすが、これらのSNP 対立遺伝子はすべて一般的であり、負の選択では精製されない[29]。 構造変異 主な記事 構造変異 構造変異とは、生物の染色体の構造の変異である。コピー数の変異や欠失、逆位、挿入、重複などの構造的変異は、ヒトの遺伝的変異を一塩基の多様性よりもは るかに多く占めている。これは2007年、2人のヒトのゲノムの2倍体全塩基配列の解析から結論づけられた: クレイグ・ベンターとジェームズ・D・ワトソンである。これは、ヒトゲノムプロジェクトとセレラ・ゲノミクスがそれぞれ発表した、多くの個体からの配列の 融合である2つのハプロイド配列に加えられたものである[30]。 1000 Genomes Projectによると、典型的なヒトには2,100から2,500の構造変異があり、これには約1,000の大きな欠失、160のコピー数変異、915 のAlu挿入、128のL1挿入、51のSVA挿入、4つのNUMT、10の逆位が含まれる[2]。 コピー数の変異 主な記事 コピー数の変異 コピー数多型変異(CNV)とは、染色体上のDNAの大きな領域が欠失または重複することによるゲノムの差異である。血縁関係のないヒトのゲノムの 0.4%がコピー数に関して異なると推定されている。コピー数の変異を含めると、ヒトとヒトの遺伝的変異は少なくとも0.5%(類似度99.5%)と推定 される[31][32][33][34]。コピー数の変異は遺伝するが、発生過程で生じることもある[35][36][37][38]。 ネアンデルタール人5万人[39]に対して、現代人参照アセンブリのゲノム変異が大きい領域を視覚的に示したマップがPratasらによって作成されている[40]。 エピジェネティクス エピジェネティックな変異とは、DNAに付着し、遺伝子がどのように読み取られるかに影響を与える化学的タグの変異である。このタグは「エピジェネティッ クなマーキングと呼ばれ、遺伝子がどのように読み取られるかを制御するスイッチとして働く」[41]。いくつかの対立遺伝子では、DNAのエピジェネ ティックな状態、および関連する表現型は、個体の世代を超えて遺伝する可能性がある[42]。 遺伝的可変性 主な記事 遺伝的変動性 遺伝的可変性とは、集団内の個々の遺伝子型が互いに異なる(異なるようになる)傾向を示す尺度である。変異性は遺伝的多様性とは異なり、特定の集団で見ら れる変異の量である。形質の変動性とは、その形質が環境や遺伝的影響に応答してどれだけ変動する傾向があるかということである。 クライン 主な記事 クライン(生物学) 生物学においてクラインとは、地理的な範囲にわたって表現型や遺伝的な差異を徐々に示す生物の種、個体群、品種、形態の連続体であり、典型的には環境の不均一性の結果として生じる。 ハプログループ 主な記事 ハプログループ 分子進化の研究において、ハプログループとは一塩基多型(SNP)変異を持つ共通の祖先を共有する類似したハプロタイプのグループのことである。ハプログループの研究は、数千年前にさかのぼる祖先の起源に関する情報を提供する[46]。 最も一般的に研究されているヒトのハプログループは、Y染色体(Y-DNA)ハプログループとミトコンドリアDNA(mtDNA)ハプログループであり、 どちらも遺伝的集団を定義するのに用いることができる。Y-DNAは父から子へと父系にのみ受け継がれ、mtDNAは母から娘または息子へと母系に受け継 がれる。Y-DNAとmtDNAは各世代で偶然の突然変異によって変化する可能性がある。 可変長タンデム反復配列 主な記事 可変反復数タンデムリピート 可変反復数タンデムリピート(VNTR)とは、タンデムリピートの長さの変化のことである。タンデムリピートとは、短い塩基配列が隣接して繰り返されるこ とである。タンデムリピートは多くの染色体に存在し、その長さは個体によって異なる。それぞれの変異体は遺伝した対立遺伝子として働くので、個人または親 の識別に使われる。タンデム反復配列の解析は、遺伝学や生物学の研究、法医学、DNA指紋採取などに有用である。 短いタンデムリピート(約5塩基対)はマイクロサテライトと呼ばれ、長いものはミニサテライトと呼ばれる。 |
History and geographic distribution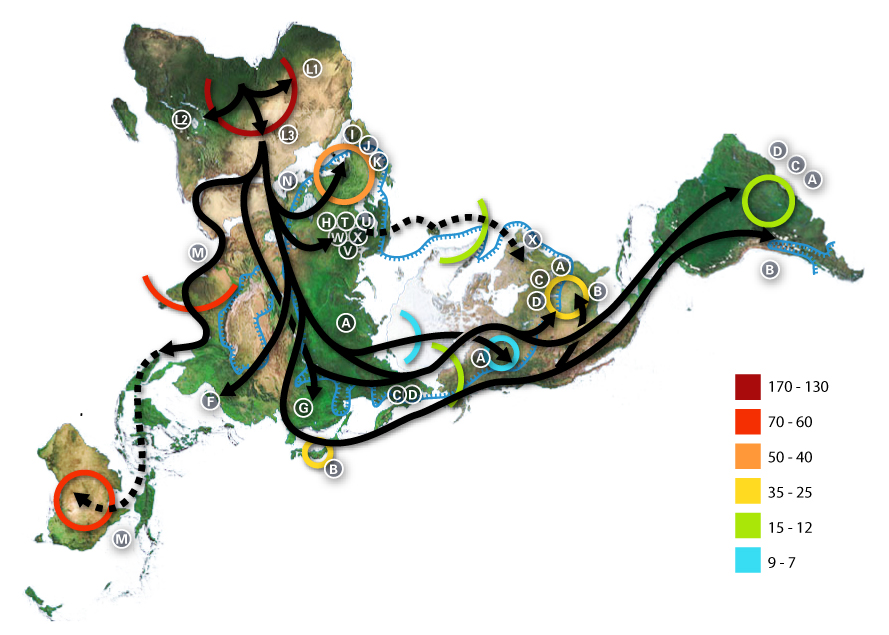 Map of the migration of modern humans out of Africa, based on mitochondrial DNA. Colored rings indicate thousand years before present. 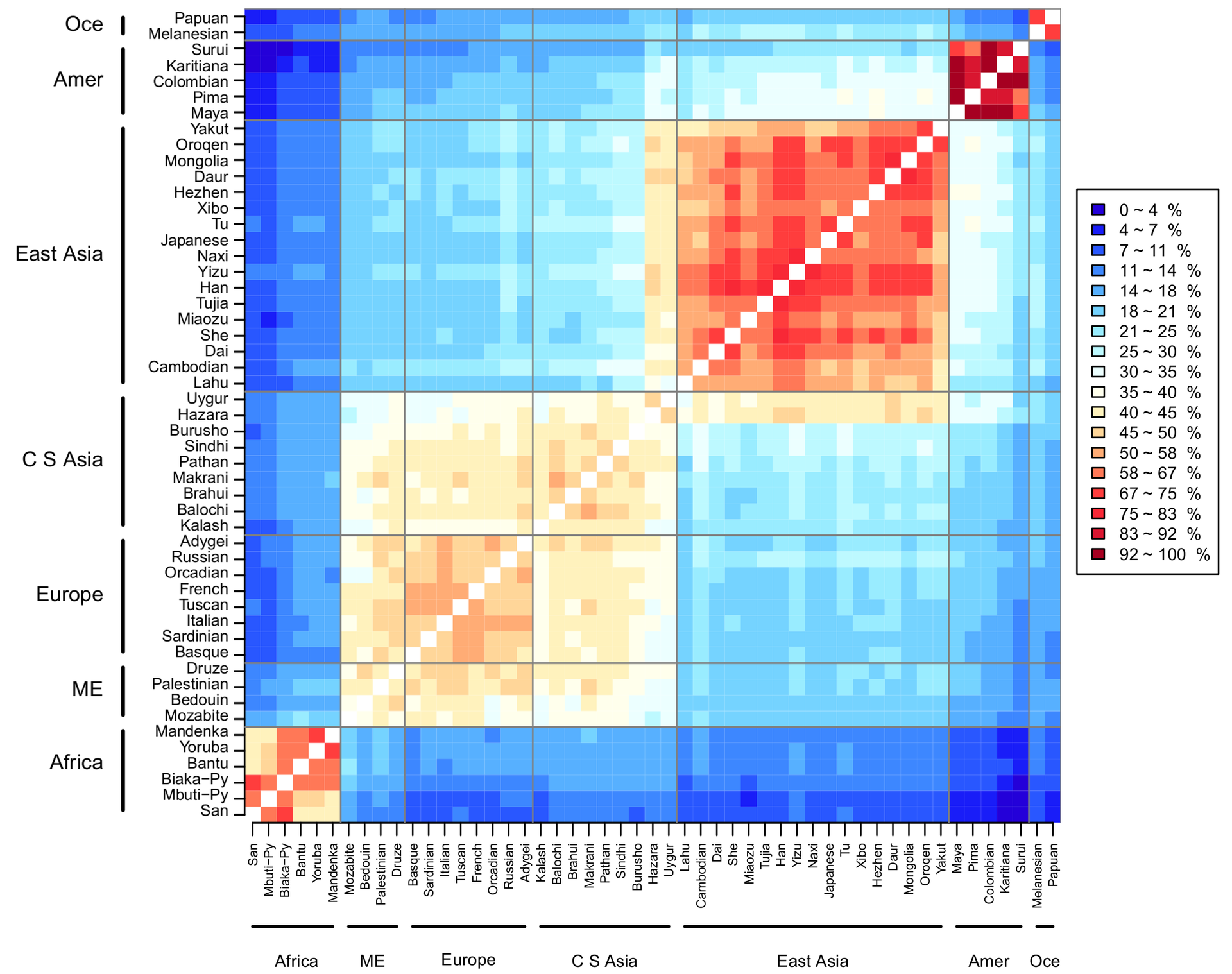 Genetic distance map by Magalhães et al. (2012) See also: Human evolutionary genetics § Modern humans, and Recent human evolution Recent African origin of modern humans The recent African origin of modern humans paradigm assumes the dispersal of non-African populations of anatomically modern humans after 70,000 years ago. Dispersal within Africa occurred significantly earlier, at least 130,000 years ago. The "out of Africa" theory originates in the 19th century, as a tentative suggestion in Charles Darwin's Descent of Man,[47] but remained speculative until the 1980s when it was supported by the study of present-day mitochondrial DNA, combined with evidence from physical anthropology of archaic specimens. According to a 2000 study of Y-chromosome sequence variation,[48] human Y-chromosomes trace ancestry to Africa, and the descendants of the derived lineage left Africa and eventually were replaced by archaic human Y-chromosomes in Eurasia. The study also shows that a minority of contemporary populations in East Africa and the Khoisan are the descendants of the most ancestral patrilineages of anatomically modern humans that left Africa 35,000 to 89,000 years ago.[48] Other evidence supporting the theory is that variations in skull measurements decrease with distance from Africa at the same rate as the decrease in genetic diversity. Human genetic diversity decreases in native populations with migratory distance from Africa, and this is thought to be due to bottlenecks during human migration, which are events that temporarily reduce population size.[49][50] A 2009 genetic clustering study, which genotyped 1327 polymorphic markers in various African populations, identified six ancestral clusters. The clustering corresponded closely with ethnicity, culture and language.[51] A 2018 whole genome sequencing study of the world's populations observed similar clusters among the populations in Africa. At K=9, distinct ancestral components defined the Afroasiatic-speaking populations inhabiting North Africa and Northeast Africa; the Nilo-Saharan-speaking populations in Northeast Africa and East Africa; the Ari populations in Northeast Africa; the Niger-Congo-speaking populations in West-Central Africa, West Africa, East Africa and Southern Africa; the Pygmy populations in Central Africa; and the Khoisan populations in Southern Africa.[52] In May 2023, scientists reported, based on genetic studies, a more complicated pathway of human evolution than previously understood. According to the studies, humans evolved from different places and times in Africa, instead of from a single location and period of time.[53][54] Population genetics See also: Population genetics Because of the common ancestry of all humans, only a small number of variants have large differences in frequency between populations. However, some rare variants in the world's human population are much more frequent in at least one population (more than 5%).[55]  Genetic variation 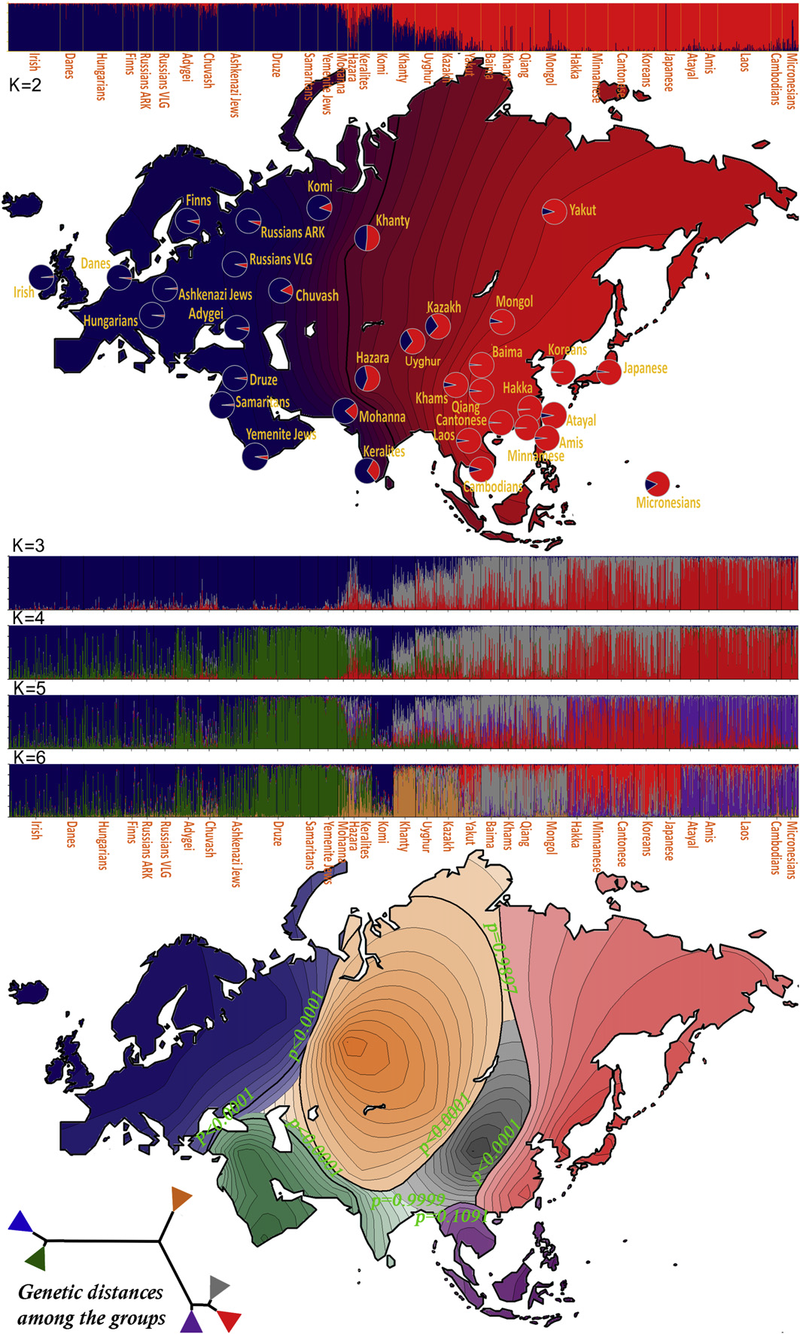 Genetic variation of Eurasian populations showing different frequency of West- and East-Eurasian components.[56] It is commonly assumed that early humans left Africa, and thus must have passed through a population bottleneck before their African-Eurasian divergence around 100,000 years ago (ca. 3,000 generations). The rapid expansion of a previously small population has two important effects on the distribution of genetic variation. First, the so-called founder effect occurs when founder populations bring only a subset of the genetic variation from their ancestral population. Second, as founders become more geographically separated, the probability that two individuals from different founder populations will mate becomes smaller. The effect of this assortative mating is to reduce gene flow between geographical groups and to increase the genetic distance between groups.[citation needed] The expansion of humans from Africa affected the distribution of genetic variation in two other ways. First, smaller (founder) populations experience greater genetic drift because of increased fluctuations in neutral polymorphisms. Second, new polymorphisms that arose in one group were less likely to be transmitted to other groups as gene flow was restricted.[citation needed] Populations in Africa tend to have lower amounts of linkage disequilibrium than do populations outside Africa, partly because of the larger size of human populations in Africa over the course of human history and partly because the number of modern humans who left Africa to colonize the rest of the world appears to have been relatively low.[57] In contrast, populations that have undergone dramatic size reductions or rapid expansions in the past and populations formed by the mixture of previously separate ancestral groups can have unusually high levels of linkage disequilibrium[57] Distribution of variation 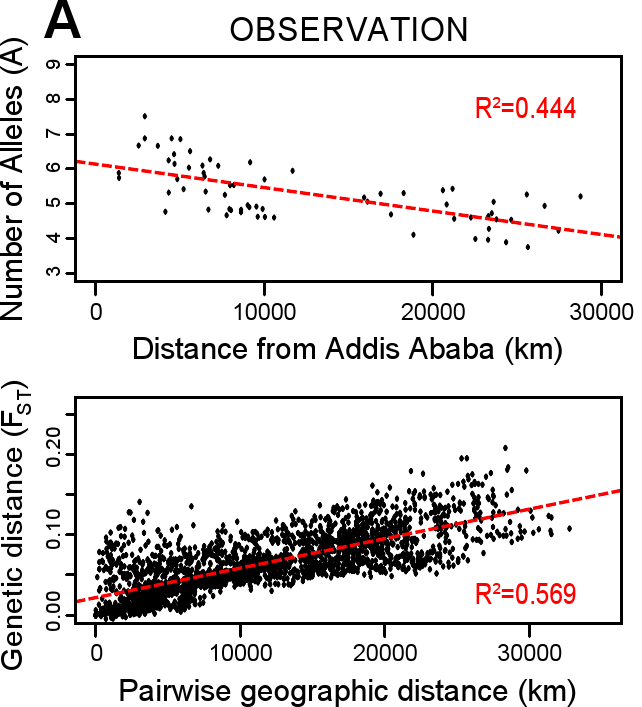 Human genetic variation calculated from genetic data representing 346 microsatellite loci taken from 1484 individuals in 78 human populations. The upper graph illustrates that as populations are further from East Africa, they have declining genetic diversity as measured in average number of microsatellite repeats at each of the loci. The bottom chart illustrates isolation by distance. Populations with a greater distance between them are more dissimilar (as measured by the Fst statistic) than those which are geographically close to one another. The horizontal axis of both charts is geographic distance as measured along likely routes of human migration. (Chart from Kanitz et al. 2018) The distribution of genetic variants within and among human populations are impossible to describe succinctly because of the difficulty of defining a "population," the clinal nature of variation, and heterogeneity across the genome (Long and Kittles 2003). In general, however, an average of 85% of genetic variation exists within local populations, ~7% is between local populations within the same continent, and ~8% of variation occurs between large groups living on different continents.[58][59] The recent African origin theory for humans would predict that in Africa there exists a great deal more diversity than elsewhere and that diversity should decrease the further from Africa a population is sampled. Phenotypic variation Further information: Phenotype § Phenotypic variation Sub-Saharan Africa has the most human genetic diversity and the same has been shown to hold true for phenotypic variation in skull form.[49][60] Phenotype is connected to genotype through gene expression. Genetic diversity decreases smoothly with migratory distance from that region, which many scientists believe to be the origin of modern humans, and that decrease is mirrored by a decrease in phenotypic variation. Skull measurements are an example of a physical attribute whose within-population variation decreases with distance from Africa. The distribution of many physical traits resembles the distribution of genetic variation within and between human populations (American Association of Physical Anthropologists 1996; Keita and Kittles 1997). For example, ~90% of the variation in human head shapes occurs within continental groups, and ~10% separates groups, with a greater variability of head shape among individuals with recent African ancestors (Relethford 2002). A prominent exception to the common distribution of physical characteristics within and among groups is skin color. Approximately 10% of the variance in skin color occurs within groups, and ~90% occurs between groups (Relethford 2002). This distribution of skin color and its geographic patterning – with people whose ancestors lived predominantly near the equator having darker skin than those with ancestors who lived predominantly in higher latitudes – indicate that this attribute has been under strong selective pressure. Darker skin appears to be strongly selected for in equatorial regions to prevent sunburn, skin cancer, the photolysis of folate, and damage to sweat glands.[61] Understanding how genetic diversity in the human population impacts various levels of gene expression is an active area of research. While earlier studies focused on the relationship between DNA variation and RNA expression, more recent efforts are characterizing the genetic control of various aspects of gene expression including chromatin states,[62] translation,[63] and protein levels.[64] A study published in 2007 found that 25% of genes showed different levels of gene expression between populations of European and Asian descent.[65][66][67][68][69] The primary cause of this difference in gene expression was thought to be SNPs in gene regulatory regions of DNA. Another study published in 2007 found that approximately 83% of genes were expressed at different levels among individuals and about 17% between populations of European and African descent.[70][71] Wright's fixation index as measure of variation The population geneticist Sewall Wright developed the fixation index (often abbreviated to FST) as a way of measuring genetic differences between populations. This statistic is often used in taxonomy to compare differences between any two given populations by measuring the genetic differences among and between populations for individual genes, or for many genes simultaneously.[72] It is often stated that the fixation index for humans is about 0.15. This translates to an estimated 85% of the variation measured in the overall human population is found within individuals of the same population, and about 15% of the variation occurs between populations. These estimates imply that any two individuals from different populations may be more similar to each other than either is to a member of their own group.[73][74] "The shared evolutionary history of living humans has resulted in a high relatedness among all living people, as indicated for example by the very low fixation index (FST) among living human populations." Richard Lewontin, who affirmed these ratios, thus concluded neither "race" nor "subspecies" were appropriate or useful ways to describe human populations.[58] Wright himself believed that values >0.25 represent very great genetic variation and that an FST of 0.15–0.25 represented great variation. However, about 5% of human variation occurs between populations within continents, therefore FST values between continental groups of humans (or races) of as low as 0.1 (or possibly lower) have been found in some studies, suggesting more moderate levels of genetic variation.[72] Graves (1996) has countered that FST should not be used as a marker of subspecies status, as the statistic is used to measure the degree of differentiation between populations,[72] although see also Wright (1978).[75] Jeffrey Long and Rick Kittles give a long critique of the application of FST to human populations in their 2003 paper "Human Genetic Diversity and the Nonexistence of Biological Races". They find that the figure of 85% is misleading because it implies that all human populations contain on average 85% of all genetic diversity. They argue the underlying statistical model incorrectly assumes equal and independent histories of variation for each large human population. A more realistic approach is to understand that some human groups are parental to other groups and that these groups represent paraphyletic groups to their descent groups. For example, under the recent African origin theory the human population in Africa is paraphyletic to all other human groups because it represents the ancestral group from which all non-African populations derive, but more than that, non-African groups only derive from a small non-representative sample of this African population. This means that all non-African groups are more closely related to each other and to some African groups (probably east Africans) than they are to others, and further that the migration out of Africa represented a genetic bottleneck, with much of the diversity that existed in Africa not being carried out of Africa by the emigrating groups. Under this scenario, human populations do not have equal amounts of local variability, but rather diminished amounts of diversity the further from Africa any population lives. Long and Kittles find that rather than 85% of human genetic diversity existing in all human populations, about 100% of human diversity exists in a single African population, whereas only about 70% of human genetic diversity exists in a population derived from New Guinea. Long and Kittles argued that this still produces a global human population that is genetically homogeneous compared to other mammalian populations.[76] Archaic admixture Main article: Archaic human admixture with modern humans Anatomically modern humans interbred with Neanderthals during the Middle Paleolithic. In May 2010, the Neanderthal Genome Project presented genetic evidence that interbreeding took place and that a small but significant portion, around 2–4%, of Neanderthal admixture is present in the DNA of modern Eurasians and Oceanians, and nearly absent in sub-Saharan African populations.[77][78] Between 4% and 6% of the genome of Melanesians (represented by the Papua New Guinean and Bougainville Islander) appears to derive from Denisovans – a previously unknown hominin which is more closely related to Neanderthals than to Sapiens. It was possibly introduced during the early migration of the ancestors of Melanesians into Southeast Asia. This history of interaction suggests that Denisovans once ranged widely over eastern Asia.[79] Thus, Melanesians emerge as one of the most archaic-admixed populations, having Denisovan/Neanderthal-related admixture of ~8%.[79] In a study published in 2013, Jeffrey Wall from University of California studied whole sequence-genome data and found higher rates of introgression in Asians compared to Europeans.[80] Hammer et al. tested the hypothesis that contemporary African genomes have signatures of gene flow with archaic human ancestors and found evidence of archaic admixture in the genomes of some African groups, suggesting that modest amounts of gene flow were widespread throughout time and space during the evolution of anatomically modern humans.[81] A study published in 2020 found that the Yoruba and Mende populations of West Africa derive between 2% and 19% of their genome from an as-yet unidentified archaic hominin population that likely diverged before the split of modern humans and the ancestors of Neanderthals and Denisovans,[82] potentially making these groups the most archaic-admixed human populations identified yet. |
歴史と地理的分布 ミトコンドリアDNAに基づく、現生人類のアフリカからの移動の地図。色のついたリングは現在から千年前を示す。 Magalhãesらによる遺伝的距離マップ(2012年) 以下も参照のこと: 人類進化遺伝学§現生人類、および最近の人類進化も参照のこと。 現生人類のアフリカ起源説 現生人類の最近のアフリカ起源パラダイムは、7万年前以降に解剖学的に現生人類の非アフリカ人集団が分散したと仮定している。アフリカ国内への拡散はそれ よりかなり早く、少なくとも13万年前には起こっていた。アフリカ外」説は19世紀にチャールズ・ダーウィンの『人間の下降』[47]の中で暫定的な提案 として生まれたが、1980年代に現在のミトコンドリアDNAの研究と古代の標本の身体人類学からの証拠とが組み合わされて支持されるまで、推測の域を出 なかった。 2000年のY染色体配列変異の研究によると[48]、ヒトのY染色体は祖先をアフリカに辿り、派生した系統の子孫はアフリカを離れ、やがてユーラシア大 陸で古代のヒトのY染色体に取って代わられた。この研究はまた、東アフリカとコイサンに住む少数派の現代人が、35,000~89,000年前にアフリカ を離れた、解剖学的に現生人類の最も祖先的な父系群の子孫であることを示している[48]。この説を支持する他の証拠としては、頭蓋骨の大きさのバリエー ションが、遺伝的多様性の減少と同じ割合で、アフリカからの距離とともに減少していることが挙げられる。ヒトの遺伝的多様性は、アフリカからの移動距離と ともに先住民の集団で減少しており、これはヒトが移動する際のボトルネック(集団規模を一時的に縮小させる事象)によるものと考えられている[49] [50]。 2009年に行われた遺伝的クラスタリング研究では、アフリカの様々な集団において1327の多型マーカーを遺伝子型決定し、6つの祖先クラスターが特定 された。このクラスタリングは民族、文化、言語と密接に対応していた[51]。2018年に行われた世界の集団の全ゲノム配列決定研究でも、アフリカの集 団の間に同様のクラスタが観察された。K=9では、北アフリカと北東アフリカに居住するアフロアシア語を話す集団、北東アフリカと東アフリカのニロ・サハ ラ語を話す集団、北東アフリカのアリ族集団、西中央アフリカ、西アフリカ、東アフリカ、南部アフリカのニジェール・コンゴ語を話す集団、中央アフリカのピ グミー族集団、南部アフリカのコイサン族集団が、明確な祖先構成要素によって定義されていた[52]。 2023年5月、科学者たちは遺伝子研究に基づき、これまで理解されていたよりも複雑な人類進化の経路を報告した。その研究によると、人類はアフリカの単一の場所と時代から進化したのではなく、アフリカの異なる場所と時代から進化したのである[53][54]。 集団遺伝学 以下も参照: 集団遺伝学 ヒトの祖先は共通であるため、集団間で頻度に大きな差がある変異体はごく少数である。しかし、世界のヒト集団の中には、少なくとも1つの集団で頻度が高い(5%以上)稀な変異体もある[55]。 遺伝的変異 西ユーラシアと東ユーラシアの成分の頻度が異なることを示すユーラシア集団の遺伝的変異[56]。 一般的に、初期の人類はアフリカを出発し、約10万年前(約3,000世代前)にアフリカとユーラシアが分岐する前に、集団ボトルネックを通過したに違い ないと考えられている。それまで少なかった集団が急速に拡大することは、遺伝的変異の分布に2つの重要な影響を与える。第一に、いわゆる創始者効果は、創 始者集団が祖先集団から遺伝的変異の一部だけを持ち込む場合に起こる。第二に、創始者が地理的に離れるにつれて、異なる創始者集団の2個体が交尾する確率 は小さくなる。このような同系交配の影響は、地理的集団間の遺伝子フローを減少させ、集団間の遺伝的距離を拡大させる。 アフリカからのヒトの拡大は、遺伝的変異の分布に他の2つの影響を与えた。第一に、小規模(創始者)集団では中立多型の変動が大きくなるため、遺伝的ドリ フトが大きくなる。第二に、遺伝子の流れが制限されたため、ある集団で生じた新しい多型が他の集団に伝わりにくくなった。 アフリカの集団はアフリカ以外の地域の集団よりも連鎖不平衡の量が少ない傾向があるが、これは人類の歴史の中でアフリカのヒト集団の規模が大きかったこと や、アフリカを出て世界の他の地域を植民地化した現生人類の数が比較的少なかったことが一因である[57]。対照的に、過去に劇的な規模の縮小や急激な拡 大を経験した集団や、以前は別々の祖先集団が混在して形成された集団は、連鎖不平衡の量が異常に多くなることがある[57]。 変異の分布 78のヒト集団の1484個体から採取した346のマイクロサテライト遺伝子座から算出したヒトの遺伝的変異。上のグラフは、各遺伝子座におけるマイクロ サテライトの平均反復数で測定したもので、東アフリカから遠くなるにつれて、集団の遺伝的多様性が減少していることを示している。下のグラフは距離による 隔離を示している。距離が離れている個体群の方が、地理的に近い個体群よりも(Fst統計で測定した)非類似度が高い。両チャートの横軸は、人類が移動し たと思われるルートに沿って測定した地理的距離である。(グラフはKanitz et al.) ヒト集団内および集団間の遺伝的変異の分布は、「集団」を定義することの難しさ、変異のクリンナルな性質、ゲノム全体の不均一性のために、簡潔に説明する ことは不可能である(Long and Kittles 2003)。しかし、一般的には、遺伝的変異の平均85%が地域集団内に存在し、7%程度が同じ大陸内の地域集団間に存在し、8%程度が異なる大陸に住む 大きな集団間に存在する[58][59]。最近のヒトのアフリカ起源説では、アフリカには他の地域よりも非常に多くの多様性が存在し、アフリカから遠く離 れた集団からサンプリングされるほど多様性は減少すると予測されている。 表現型の変異 さらなる情報 表現型§表現型の変異 サハラ以南のアフリカはヒトの遺伝的多様性が最も高く、頭蓋骨の形の表現型の変異についても同様であることが示されている[49][60]。遺伝的多様性 は、多くの科学者が現生人類の起源であると考えているその地域からの移動距離とともに滑らかに減少し、その減少は表現型の変異の減少によって反映される。 頭蓋骨の大きさは、集団内の変異がアフリカからの距離とともに減少する身体的特徴の一例である。 多くの身体的特徴の分布は、人類集団内および集団間の遺伝的変異の分布に似ている(American Association of Physical Anthropologists 1996; Keita and Kittles 1997)。例えば、ヒトの頭部形状の変異の約90%は大陸グループ内で、約10%はグループ間で生じており、最近のアフリカ人を祖先に持つ個体では頭部 形状の変異が大きい(Relethford 2002)。 集団内および集団間の身体的特徴の共通分布の顕著な例外は、皮膚の色である。肌の色のばらつきの約10%が集団内で、約90%が集団間で生じている (Relethford 2002年)。このような肌の色の分布とその地理的パターン(祖先が主に赤道付近に住んでいた人々は、祖先が主に高緯度に住んでいた人々よりも肌が黒い) は、この属性が強い選択圧を受けてきたことを示している。赤道直下の地域では、日焼け、皮膚がん、葉酸の光分解、汗腺の損傷を防ぐために、肌が黒いことが 強く選択されているようである[61]。 ヒト集団における遺伝的多様性が、様々なレベルの遺伝子発現にどのような影響を与えるかを理解することは、活発な研究分野である。初期の研究ではDNAの 変異とRNA発現の関係に焦点が当てられていたが、最近では、クロマチン状態[62]、翻訳[63]、タンパク質レベルなど、遺伝子発現の様々な側面の遺 伝的制御を特徴づける研究が進められている[64]。2007年に発表された研究では、遺伝子の25%がヨーロッパ系とアジア系の集団間で遺伝子発現のレ ベルに差があることが判明した[65][66][67][68][69]。この遺伝子発現の差の主な原因は、DNAの遺伝子制御領域におけるSNPである と考えられていた。2007年に発表された別の研究では、遺伝子の約83%が個人間で異なるレベルで発現しており、ヨーロッパ系とアフリカ系の集団間では 約17%であった[70][71]。 変異の尺度としてのライトの固定指数 集団遺伝学者のSewall Wrightは集団間の遺伝的差異を測定する方法として固定指数(しばしばFSTと略される)を開発した。この統計量は分類学において、個々の遺伝子につ いて、あるいは多くの遺伝子について、集団間や集団間の遺伝的差異を同時に測定することによって、任意の2つの集団間の差異を比較するためによく使われる [72]。ヒトの固定化指数は約0.15であるとよく言われる。これは、ヒトの集団全体で測定された変異の85%が同じ集団の個体内にあり、約15%が集 団間で生じていると推定されることを意味する。73][74]「現生人類は進化の歴史を共有しているため、現生人類集団間の固定指数(FST)が非常に低 いことなどが示すように、現生人類間の血縁性が高い。これらの比率を肯定したリチャード・レウォンティンは、このように「人種」も「亜種」もヒト集団を記 述するのに適切でも有用でもないと結論づけた[58]。 ライト自身は、0.25を超える値は非常に大きな遺伝的変異を表し、0.15-0.25のFSTは大きな変異を表すと考えていた。しかし、ヒトの変異の約 5%は大陸内の個体群間で生じているため、大陸のヒトのグループ(または人種)間のFST値は0.1(またはそれ以下かもしれない)という低い値がいくつ かの研究で見つかっており、遺伝的変異がより中程度のレベルであることを示唆している[72]。Graves(1996)は、FSTは個体群間の分化の程 度を測定するために使用される統計であるため、亜種ステータスのマーカーとして使用すべきではないと反論している[72]が、Wright(1978)も 参照のこと[75]。 ジェフリー・ロングとリック・キトルスは2003年の論文 「Human Genetic Diversity and the Nonxistence of Biological Races(ヒトの遺伝的多様性と生物学的種族の非存在)」の中で、ヒト集団へのFSTの適用について長い批判を行っている。彼らは、85%という数字 は、すべてのヒト集団が遺伝的多様性の平均85%を含んでいることを意味するため、誤解を招きかねないとしている。この統計モデルは、各人類集団の変異の 歴史が平等で独立したものであると誤って仮定している。より現実的なアプローチとしては、あるヒト集団は他の集団の親であり、これらの集団はその子孫集団 に対してパラ系統集団であると理解することである。例えば、最近のアフリカ起源説の下では、アフリカの人類集団は他のすべての人類集団に対してパラファイ ト的である。なぜなら、アフリカの人類集団は、すべての非アフリカ人集団の祖先集団であるが、それ以上に、非アフリカ人集団は、このアフリカ人集団の小さ な代表的でないサンプルに由来するだけだからである。つまり、すべての非アフリカ人集団は、他の集団よりも互いに、また一部のアフリカ人集団(おそらく東 アフリカ人)により近縁であり、さらにアフリカからの移住は遺伝的ボトルネックであり、アフリカに存在した多様性の多くは、移住集団によってアフリカから 持ち出されなかったということである。このシナリオのもとでは、人間の集団は局所的な多様性を等しく持っているのではなく、アフリカから遠く離れれば離れ るほど多様性が減少していくことになる。ロングとキトルスは、ヒトの遺伝的多様性の85%が全人類集団に存在するのではなく、ヒトの多様性の約100%が アフリカの単一集団に存在するのに対し、ニューギニアに由来する集団にはヒトの遺伝的多様性の約70%しか存在しないことを発見した。ロングとキトルス は、これでもなお、他の哺乳類集団と比較して遺伝的に均質な世界的なヒト集団を生み出していると主張した[76]。 古代の混血 主な記事 古人類と現生人類の混血 解剖学的に現生人類は中期旧石器時代にネアンデルタール人と混血した。2010年5月、ネアンデルタール人ゲノム・プロジェクトは、交雑が行われ、現代の ユーラシア人とオセアニア人のDNAには、ネアンデルタール人との混血がわずかではあるが2~4%程度存在し、サハラ以南のアフリカの集団にはほとんど見 られないという遺伝学的証拠を発表した[77][78]。 メラネシア人(パプアニューギニア人とブーゲンビル島民に代表される)のゲノムの4%から6%はデニソワ人に由来するようである。デニソワ人はメラネシア 人の祖先が東南アジアに移動した初期に持ち込まれた可能性がある。このような相互作用の歴史は、デニソワ人がかつて東アジアに広く分布していたことを示唆 している[79]。 このように、メラネシア人はデニソワ人とネアンデルタール人関連の混血が〜8%であり、最も古風な混血集団の一つとして浮かび上がってくる[79]。 2013年に発表された研究では、カリフォルニア大学のジェフリー・ウォールが全塩基配列-ゲノムデータを調査し、ヨーロッパ人と比較してアジア人におけ る内挿率が高いことを発見した[80]。ハマーらは、現代のアフリカ人ゲノムには古代の人類の祖先との遺伝子流動のサインがあるという仮説を検証し、いく つかのアフリカ人グループのゲノムに古代の混血の証拠を発見した。 2020年に発表された研究によると、西アフリカのヨルバとメンデの集団は、ゲノムの2%から19%が、現生人類とネアンデルタール人やデニソワ人の祖先 が分岐する前に分岐したと思われる、まだ確認されていない古人類集団に由来していることが判明しており[82]、これらの集団は、現在確認されている中で 最も古人類と混血したヒト集団となる可能性がある。 |
Categorization of the world population Chart showing human genetic clustering[83]  Individuals mostly have genetic variants which are found in multiple regions of the world. Based on data from "A unified genealogy of modern and ancient genomes".[84] See also: Race (human classification) and Race and genetics New data on human genetic variation has reignited the debate about a possible biological basis for categorization of humans into races. Most of the controversy surrounds the question of how to interpret the genetic data and whether conclusions based on it are sound. Some researchers argue that self-identified race can be used as an indicator of geographic ancestry for certain health risks and medications. Although the genetic differences among human groups are relatively small, these differences in certain genes such as duffy, ABCC11, SLC24A5, called ancestry-informative markers (AIMs) nevertheless can be used to reliably situate many individuals within broad, geographically based groupings. For example, computer analyses of hundreds of polymorphic loci sampled in globally distributed populations have revealed the existence of genetic clustering that roughly is associated with groups that historically have occupied large continental and subcontinental regions (Rosenberg et al. 2002; Bamshad et al. 2003). Some commentators have argued that these patterns of variation provide a biological justification for the use of traditional racial categories. They argue that the continental clusterings correspond roughly with the division of human beings into sub-Saharan Africans; Europeans, Western Asians, Central Asians, Southern Asians and Northern Africans; Eastern Asians, Southeast Asians, Polynesians and Native Americans; and other inhabitants of Oceania (Melanesians, Micronesians & Australian Aborigines) (Risch et al. 2002). Other observers disagree, saying that the same data undercut traditional notions of racial groups (King and Motulsky 2002; Calafell 2003; Tishkoff and Kidd 2004[24]). They point out, for example, that major populations considered races or subgroups within races do not necessarily form their own clusters. Racial categories are also undermined by findings that genetic variants which are limited to one region tend to be rare within that region, variants that are common within a region tend to be shared across the globe, and most differences between individuals, whether they come from the same region or different regions, are due to global variants.[85] No genetic variants have been found which are fixed within a continent or major region and found nowhere else.[86] Furthermore, because human genetic variation is clinal, many individuals affiliate with two or more continental groups. Thus, the genetically based "biogeographical ancestry" assigned to any given person generally will be broadly distributed and will be accompanied by sizable uncertainties (Pfaff et al. 2004). In many parts of the world, groups have mixed in such a way that many individuals have relatively recent ancestors from widely separated regions. Although genetic analyses of large numbers of loci can produce estimates of the percentage of a person's ancestors coming from various continental populations (Shriver et al. 2003; Bamshad et al. 2004), these estimates may assume a false distinctiveness of the parental populations, since human groups have exchanged mates from local to continental scales throughout history (Cavalli-Sforza et al. 1994; Hoerder 2002). Even with large numbers of markers, information for estimating admixture proportions of individuals or groups is limited, and estimates typically will have wide confidence intervals (Pfaff et al. 2004). Genetic clustering Main article: Human genetic clustering Genetic data can be used to infer population structure and assign individuals to groups that often correspond with their self-identified geographical ancestry. Jorde and Wooding (2004) argued that "Analysis of many loci now yields reasonably accurate estimates of genetic similarity among individuals, rather than populations. Clustering of individuals is correlated with geographic origin or ancestry."[23] However, identification by geographic origin may quickly break down when considering historical ancestry shared between individuals back in time.[87] An analysis of autosomal SNP data from the International HapMap Project (Phase II) and CEPH Human Genome Diversity Panel samples was published in 2009. The study of 53 populations taken from the HapMap and CEPH data (1138 unrelated individuals) suggested that natural selection may shape the human genome much more slowly than previously thought, with factors such as migration within and among continents more heavily influencing the distribution of genetic variations.[88] A similar study published in 2010 found strong genome-wide evidence for selection due to changes in ecoregion, diet, and subsistence particularly in connection with polar ecoregions, with foraging, and with a diet rich in roots and tubers.[89] In a 2016 study, principal component analysis of genome-wide data was capable of recovering previously-known targets for positive selection (without prior definition of populations) as well as a number of new candidate genes.[90] Forensic anthropology Forensic anthropologists can assess the ancestry of skeletal remains by analyzing skeletal morphology as well as using genetic and chemical markers, when possible.[91] While these assessments are never certain, the accuracy of skeletal morphology analyses in determining true ancestry has been estimated at 90%.[92]  Ternary plot showing average admixture of five North American ethnic groups. Individuals that self-identify with each group can be found at many locations on the map, but on average groups tend to cluster differently. Gene flow and admixture Main article: Gene flow Gene flow between two populations reduces the average genetic distance between the populations, only totally isolated human populations experience no gene flow and most populations have continuous gene flow with other neighboring populations which create the clinal distribution observed for most genetic variation. When gene flow takes place between well-differentiated genetic populations the result is referred to as "genetic admixture". Admixture mapping is a technique used to study how genetic variants cause differences in disease rates between population.[93] Recent admixture populations that trace their ancestry to multiple continents are well suited for identifying genes for traits and diseases that differ in prevalence between parental populations. African-American populations have been the focus of numerous population genetic and admixture mapping studies, including studies of complex genetic traits such as white cell count, body-mass index, prostate cancer and renal disease.[94] An analysis of phenotypic and genetic variation including skin color and socio-economic status was carried out in the population of Cape Verde which has a well documented history of contact between Europeans and Africans. The studies showed that pattern of admixture in this population has been sex-biased (involving mostly matings between European men and African women) and there is a significant interaction between socioeconomic status and skin color, independent of ancestry.[95] Another study shows an increased risk of graft-versus-host disease complications after transplantation due to genetic variants in human leukocyte antigen (HLA) and non-HLA proteins.[96] |
世界人口の分類 ヒトの遺伝子クラスターを示す図[83]。 ほとんどの個体は、世界の複数の地域で見られる遺伝子変異を持っている。A unified genealogy of modern and ancient genomes」のデータに基づく[84]。 以下も参照のこと: 人種(人間の分類)、人種と遺伝学 ヒトの遺伝的変異に関する新しいデータは、ヒトを人種に分類するための生物学的根拠の可能性に関する議論を再燃させている。論争の大半は、遺伝子データを どのように解釈するか、またそれに基づく結論が正しいかどうかという問題を取り巻いている。研究者の中には、特定の健康リスクや薬について、自認する人種 を地理的な祖先の指標として使うことができると主張する者もいる。 ヒト集団間の遺伝的差異は比較的小さいが、ダッフィー、ABCC11、SLC24A5などの特定の遺伝子におけるこれらの差異は、祖先情報マーカー (AIM)と呼ばれ、それにもかかわらず、多くの個人を地理的に基づいた広範な集団の中に確実に位置づけるために使用することができる。例えば、世界的に 分布する個体群からサンプリングした数百の多型遺伝子座をコンピューター解析した結果、歴史的に大陸や亜大陸の広い地域を占めてきた集団とおおよそ関連す る遺伝的クラスターが存在することが明らかになった(Rosenbergら 2002; Bamshadら 2003)。 こうした変異パターンが、伝統的な人種分類の使用を生物学的に正当化すると主張する論者もいる。彼らは、大陸ごとの分類は、人類をサハラ以南のアフリカ 人、ヨーロッパ人、西アジア人、中央アジア人、南アジア人、北アフリカ人、東アジア人、東南アジア人、ポリネシア人、アメリカ先住民、オセアニアのその他 の住民(メラネシア人、ミクロネシア人、オーストラリア原住民)に分けることとほぼ一致すると主張している(Risch et al.) また、同じデータが従来の人種集団の概念を根底から覆すものであるとして、異論を唱える見解もある(King and Motulsky 2002; Calafell 2003; Tishkoff and Kidd 2004[24])。たとえば、人種や人種内のサブグループと考えられている主要な集団は、必ずしも独自のクラスターを形成しているわけではないと指摘し ている。 人種分類はまた、ある地域に限定される遺伝的変異体はその地域内では稀である傾向があり、ある地域内で共通する変異体は世界中で共有される傾向があり、同 じ地域出身であれ異なる地域出身であれ、個人間の差のほとんどはグローバルな変異体によるものである[85]という知見によっても損なわれている。ある大 陸や主要地域内で固定され、それ以外の場所では見られない遺伝的変異体は見つかっていない[86]。 さらに、ヒトの遺伝的変異は氏族的なものであるため、多くの個体が2つ以上の大陸グループに属している。そのため、遺伝学的に「生物地理学的祖先」とされる人物は、一般的に広範に分布し、かなりの不確実性を伴うことになる(Pfaff et al.) 世界の多くの地域では、多くの個体が遠く離れた地域の比較的最近の祖先を持つように、集団が混ざり合っている。大量の遺伝子座を用いた遺伝学的解析によっ て、ある人の祖先が様々な大陸集団から来た割合の推定値を出すことができるが(Shriver et al. 大量のマーカーがあっても、個体や集団の混血比率を推定するための情報は限られており、推定値は通常広い信頼区間を持つことになる(Pfaff et al.) 遺伝的クラスタリング 主な記事 ヒトの遺伝的クラスタリング 遺伝学的データは、集団構造を推測し、しばしば自認する地理的祖先と一致する集団に個体群を割り当てるために利用できる。JordeとWooding (2004)は、「多くの遺伝子座を解析することで、個体群ではなく、個体間の遺伝的類似性を合理的に正確に推定できるようになった」と論じている。しか し、地理的起源による同定は、過去に遡って個人間で共有された歴史的祖先を考慮すると、すぐに破綻する可能性がある[87]。 International HapMap Project (Phase II)とCEPH Human Genome Diversity Panelのサンプルから得られた常染色体SNPデータの解析が2009年に発表された。HapMapとCEPHのデータから抽出された53の集団 (1138人の無関係な個体)を対象としたこの研究では、自然選択はこれまで考えられていたよりもはるかにゆっくりとヒトゲノムを形成し、大陸内や大陸間 の移動などの要因が遺伝的変異の分布により大きく影響している可能性が示唆された[88]。 [88]2010年に発表された同様の研究では、生態地域、食事、生業の変化による選択について、特に極地の生態地域、採食、根や塊茎を多く含む食事との 関連において、ゲノムワイドに強い証拠を発見した[89]。2016年の研究では、ゲノムワイドデータの主成分分析によって、(事前に集団を定義すること なく)これまで知られていた正の選択のターゲットや、多くの新しい候補遺伝子を回収することができた[90]。 法人類学 法人類学者は、可能であれば遺伝子マーカーや化学マーカーを使用するだけでなく、骨格形態を分析することによって、骨格遺体の祖先を評価することができる [91]。これらの評価は決して確実なものではないが、真の祖先を決定する上での骨格形態分析の精度は90%と推定されている[92]。 北米の5つの民族グループの平均混血度を示す3元プロット。各グループを自認する個人は地図上の多くの場所に存在するが、平均的にグループは異なる場所に集まる傾向がある。 遺伝子流動と混血 主な記事 遺伝子流動 2つの集団間の遺伝子流動は集団間の平均遺伝的距離を縮めるが、完全に孤立したヒト集団だけが遺伝子流動を経験しない。十分に分化した遺伝集団間で遺伝子流動が起こると、その結果は「遺伝的混血」と呼ばれる。 混血マッピングは、遺伝的変異が集団間の疾患発生率の違いをどのように引き起こすかを研究するために使用される手法である[93]。複数の大陸に祖先を持 つ最近の混血集団は、親集団間で有病率が異なる形質や疾患の遺伝子を同定するのに適している。アフリカ系アメリカ人の集団は、白血球数、体格指数、前立腺 がん、腎疾患などの複雑な遺伝形質の研究など、多くの集団遺伝学的研究や混血マッピング研究の焦点となっている[94]。 肌の色や社会経済的地位などの表現型と遺伝的変異の分析が、ヨーロッパ人とアフリカ人の接触の歴史がよく記録されているカーボベルデの集団で実施された。 その結果、この集団における混血のパターンは性に偏りがあり(ほとんどがヨーロッパ人男性とアフリカ人女性との交配)、社会経済的地位と肌の色との間に は、祖先とは無関係に有意な相互作用があることが示された [95] 。別の研究では、ヒト白血球抗原(HLA)および非HLAタンパク質の遺伝子変異に起因する移植後の移植片対宿主病合併症のリスク増加が示されている [96] 。 |
| Health See also: Race and health Differences in allele frequencies contribute to group differences in the incidence of some monogenic diseases, and they may contribute to differences in the incidence of some common diseases.[97] For the monogenic diseases, the frequency of causative alleles usually correlates best with ancestry, whether familial (for example, Ellis–Van Creveld syndrome among the Pennsylvania Amish), ethnic (Tay–Sachs disease among Ashkenazi Jewish populations), or geographical (hemoglobinopathies among people with ancestors who lived in malarial regions). To the extent that ancestry corresponds with racial or ethnic groups or subgroups, the incidence of monogenic diseases can differ between groups categorized by race or ethnicity, and health-care professionals typically take these patterns into account in making diagnoses.[98] Even with common diseases involving numerous genetic variants and environmental factors, investigators point to evidence suggesting the involvement of differentially distributed alleles with small to moderate effects. Frequently cited examples include hypertension (Douglas et al. 1996), diabetes (Gower et al. 2003), obesity (Fernandez et al. 2003), and prostate cancer (Platz et al. 2000). However, in none of these cases has allelic variation in a susceptibility gene been shown to account for a significant fraction of the difference in disease prevalence among groups, and the role of genetic factors in generating these differences remains uncertain (Mountain and Risch 2004). Some other variations on the other hand are beneficial to human, as they prevent certain diseases and increase the chance to adapt to the environment. For example, mutation in CCR5 gene that protects against AIDS. CCR5 gene is absent on the surface of cell due to mutation. Without CCR5 gene on the surface, there is nothing for HIV viruses to grab on and bind into. Therefore, the mutation on CCR5 gene decreases the chance of an individual's risk with AIDS. The mutation in CCR5 is also quite common in certain areas, with more than 14% of the population carry the mutation in Europe and about 6–10% in Asia and North Africa.[99] 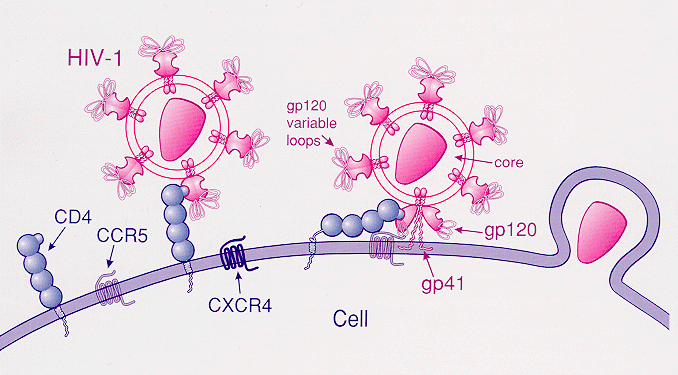 HIV attachment Apart from mutations, many genes that may have aided humans in ancient times plague humans today. For example, it is suspected that genes that allow humans to more efficiently process food are those that make people susceptible to obesity and diabetes today.[100] Neil Risch of Stanford University has proposed that self-identified race/ethnic group could be a valid means of categorization in the US for public health and policy considerations.[101][97] A 2002 paper by Noah Rosenberg's group makes a similar claim: "The structure of human populations is relevant in various epidemiological contexts. As a result of variation in frequencies of both genetic and nongenetic risk factors, rates of disease and of such phenotypes as adverse drug response vary across populations. Further, information about a patient's population of origin might provide health care practitioners with information about risk when direct causes of disease are unknown."[102] However, in 2018 Noah Rosenberg released a study arguing against genetically essentialist ideas of health disparities between populations stating environmental variants are a more likely cause[103] |
健康 こちらも参照のこと: 人種と健康 対立遺伝子頻度の差は、いくつかの単発性疾患の発生率における集団差に寄与しており、いくつかの一般的な疾患の発生率の差にも寄与している可能性がある。 [97] 単発性疾患の場合、原因対立遺伝子の頻度は、家族性(例えば、ペンシルバニアアーミッシュのエリス-ヴァンクレヴェルド症候群)、民族性(アシュケナージ ユダヤ人の集団のテイ-サックス病)、地理的(マラリア地域に住んでいた祖先を持つ人々のヘモグロビン血症)のいずれであっても、通常、祖先と最もよく相 関する。祖先が人種または民族のグループまたはサブグループに対応する限り、単発性疾患の発生率は、人種または民族によって分類されるグループ間で異なる 可能性があり、医療専門家は通常、診断を下す際にこれらのパターンを考慮に入れる[98]。 多数の遺伝的変異と環境因子が関与する一般的な疾患であっても、研究者は、影響が小さいか中等度である分布の異なる対立遺伝子の関与を示唆する証拠を指摘 している。よく引用される例としては、高血圧(Douglasら 1996年)、糖尿病(Gowerら 2003年)、肥満(Fernandezら 2003年)、前立腺がん(Platzら 2000年)などがある。しかし、いずれの場合も、疾患感受性遺伝子の対立遺伝子の変異が、グループ間の疾患有病率の差のかなりの部分を占めていることは 示されておらず、これらの差を生み出す遺伝的要因の役割は不明確なままである(Mountain and Risch 2004)。 一方、他の変異の中には、特定の疾患を予防し、環境に適応する機会を増やすという、ヒトにとって有益なものもある。例えば、エイズを予防するCCR5遺伝 子の変異である。CCR5遺伝子は変異により細胞表面に存在しない。細胞表面にCCR5遺伝子がなければ、HIVウイルスが掴んで結合するものは何もな い。従って、CCR5遺伝子の変異はAIDSのリスクを減少させる。CCR5の変異は特定の地域ではかなり一般的であり、ヨーロッパでは人口の14%以 上、アジアと北アフリカでは約6~10%が変異を持っている[99]。 HIVの付着 突然変異とは別に、太古の人類を助けたと思われる多くの遺伝子が、現代の人類を苦しめている。例えば、人間が食物をより効率的に処理できるようにする遺伝子が、今日の人々を肥満や糖尿病にしやすくしているのではないかと疑われている[100]。 スタンフォード大学のニール・リッシュ(Neil Risch)は、アメリカでは自認する人種/民族集団が、公衆衛生や政策を考慮する上で有効な分類手段となりうると提唱している[101][97]。 ノア・ローゼンバーグ(Noah Rosenberg)のグループによる2002年の論文でも、同様の主張がなされている:「ヒト集団の構造は、様々な疫学的文脈に関連している。遺伝的危 険因子と非遺伝的危険因子の頻度にばらつきがある結果、疾患や薬物有害反応のような表現型の発生率は集団によって異なる。さらに、患者の出身集団に関する 情報は、疾患の直接的な原因が不明である場合に、医療従事者にリスクに関する情報を提供する可能性がある」[102]。しかし、2018年にNoah Rosenbergは、集団間の健康格差に関する遺伝的本質主義的な考え方に反論する研究を発表し、環境変異がより可能性の高い原因であると述べている [103]。 |
| Genome projects Further information: Category:Human genome projects Human genome projects are scientific endeavors that determine or study the structure of the human genome. The Human Genome Project was a landmark genome project. |
ゲノムプロジェクト さらに詳しい情報 Category:ヒトゲノムプロジェクト ヒトゲノム計画とは、ヒトゲノムの構造を決定または研究する科学的な試みである。ヒトゲノム計画は、画期的なゲノムプロジェクトである。 |
| Archaeogenetics Chimera (genetics) Genealogical DNA test Human evolutionary genetics Isolation by distance Multiregional hypothesis Neurodiversity Race and genetics Recent single origin hypothesis Y-chromosome haplogroups in populations of the world Regional 1000 Genomes Project African admixture in Europe Genetic history of Europe Genetic history of indigenous peoples of the Americas Genetic history of South Asia Genetic history of the British Isles Projects Human Variome Project |
考古学 キメラ(遺伝学) 系図DNA検査 人類進化遺伝学 距離による隔離 多地域仮説 神経多様性 人種と遺伝学 最近の単一起源仮説 世界の集団におけるY染色体ハプログループ 地域 1000人ゲノムプロジェクト ヨーロッパにおけるアフリカ人との混血 ヨーロッパの遺伝史 アメリカ大陸先住民の遺伝史 南アジアの遺伝史 イギリス諸島の遺伝史 プロジェクト ヒト・バリオーム・プロジェクト |
| https://en.wikipedia.org/wiki/Human_genetic_variation |
|
リ ンク
文 献
そ の他の情報
Copyleft, CC, Mitzub'ixi Quq Chi'j, 1996-2099
![]()
☆
 ☆
☆