
分子人類学
Molecular anthropology
☆分子人類学(Molecular
anthropology)
は、遺伝人類学(genetic
anthropology)としても知られ、分子生物学が人類の進化の理解にどのように寄与してきたかを研究する学問である。この人類学の分野では、古代
の人類集団と現代の人類集団、また現代の種族間の進化のつながりを調べる。一般的には、DNA配列やタンパク質配列といった配列間の比較が行われるが、初
期の研究では比較血清学が用いられた。
異なる集団のDNA配列を調べることで、科学者は集団間(あるいは集団内)の関係の近さを判断することができる。遺伝的構成におけるある種の類似性によっ
て、分子人類学者は異なる集団が同じハプログループに属しているかどうか、つまり地理的起源を共有しているかどうかを判断することができる。このことは、
人類学者が移動と定住のパターンを追跡することを可能にし、現代の集団がどのように形成され、時間の経過とともに進歩してきたかについて有益な洞察を与え
るという点で重要である。
分子人類学は、チンパンジーやゴリラのような近縁種を含む、ヒトと他の霊長類の進化系統を確立するのに非常に役立っている。例えば、ヒトとチンパンジーに
は明らかに多くの形態学的類似点があるが、ある研究では、両種のDNAにはおよそ98パーセントの共通性があると結論付けている。[このような情報は、共
通の祖先を探し、人類がどのように進化してきたかをより深く理解する上で有用である。
| Molecular
anthropology, also known as genetic anthropology, is the study of how
molecular biology has contributed to the understanding of human
evolution.[1] This field of anthropology examines evolutionary links
between ancient and modern human populations, as well as between
contemporary species. Generally, comparisons are made between
sequences, either DNA or protein sequences; however, early studies used
comparative serology. By examining DNA sequences in different populations, scientists can determine the closeness of relationships between populations (or within populations). Certain similarities in genetic makeup let molecular anthropologists determine whether or not different groups of people belong to the same haplogroup, and thus if they share a common geographical origin. This is significant because it allows anthropologists to trace patterns of migration and settlement, which gives helpful insight as to how contemporary populations have formed and progressed over time.[2] Molecular anthropology has been extremely useful in establishing the evolutionary tree of humans and other primates, including closely related species like chimps and gorillas. While there are clearly many morphological similarities between humans and chimpanzees, for example, certain studies also have concluded that there is roughly a 98 percent commonality between the DNA of both species. [citation needed] However, more recent studies have modified the commonality of 98 percent to a commonality of 94 percent, showing that the genetic gap between humans and chimps is larger than originally thought.[3] Such information is useful in searching for common ancestors and coming to a better understanding of how humans evolved. |
分子人類学は、遺伝人類学としても知られ、分子生物学が人類の進化の理
解にどのように寄与してきたかを研究する学問である[1]。この人類学の分野では、古代の人類集団と現代の人類集団、また現代の種族間の進化のつながりを
調べる。一般的には、DNA配列やタンパク質配列といった配列間の比較が行われるが、初期の研究では比較血清学が用いられた。 異なる集団のDNA配列を調べることで、科学者は集団間(あるいは集団内)の関係の近さを判断することができる。遺伝的構成におけるある種の類似性によっ て、分子人類学者は異なる集団が同じハプログループに属しているかどうか、つまり地理的起源を共有しているかどうかを判断することができる。このことは、 人類学者が移動と定住のパターンを追跡することを可能にし、現代の集団がどのように形成され、時間の経過とともに進歩してきたかについて有益な洞察を与え るという点で重要である[2]。 分子人類学は、チンパンジーやゴリラのような近縁種を含む、ヒトと他の霊長類の進化系統を確立するのに非常に役立っている。例えば、ヒトとチンパンジーに は明らかに多くの形態学的類似点があるが、ある研究では、両種のDNAにはおよそ98パーセントの共通性があると結論付けている。[このような情報は、共 通の祖先を探し、人類がどのように進化してきたかをより深く理解する上で有用である。 |
Haploid loci in molecular anthropology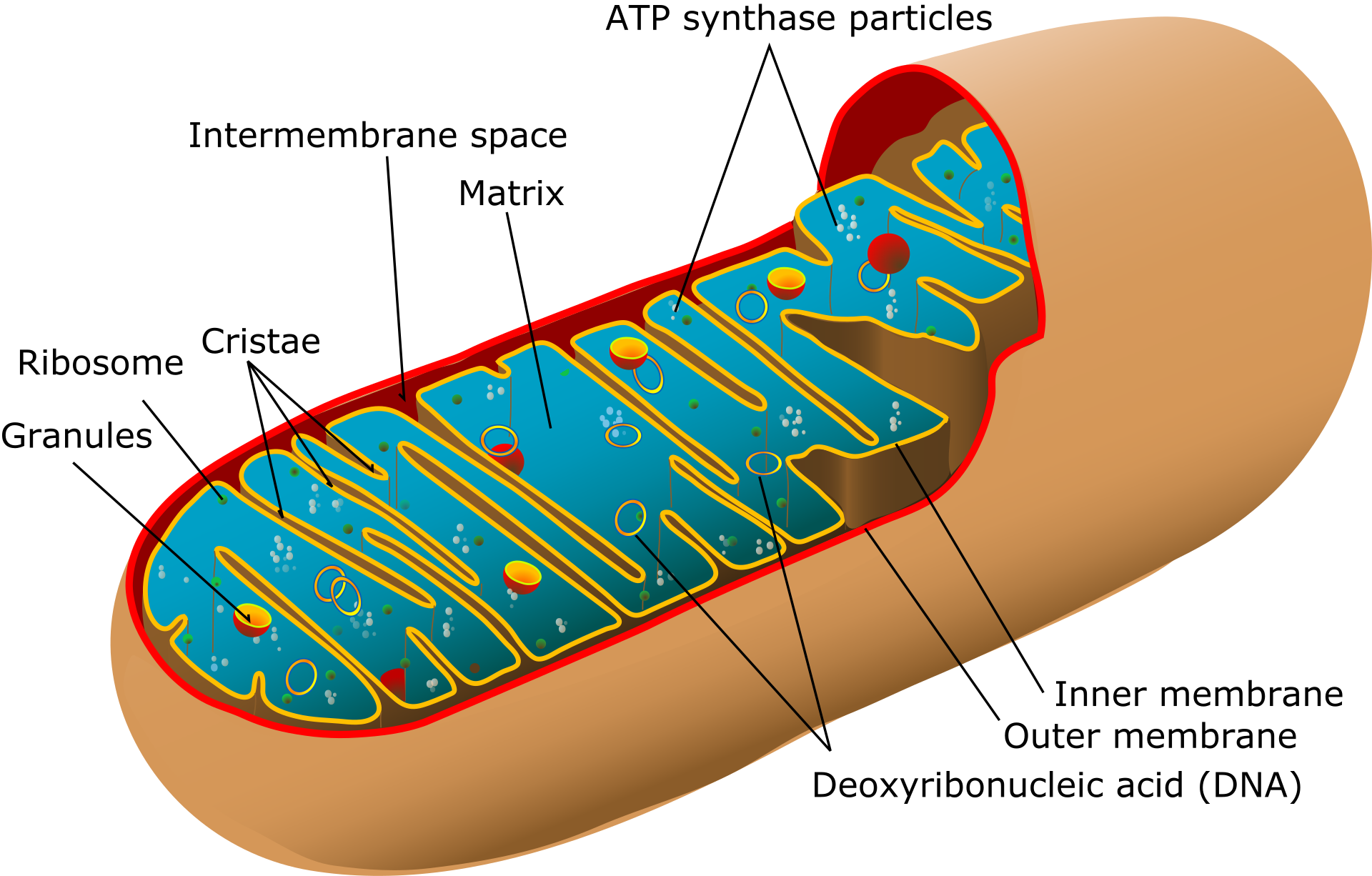 Image of mitochondrion. There are many mitochondria within a cell, and DNA in them replicates independently of the chromosomes in the nucleus. There are two continuous linkage groups in humans that are carried by a single sex. The first is the Y chromosome, which is passed from father to son. Anatomical females carry a Y chromosome only rarely, as a result of genetic defect. The other linkage group is the mitochondrial DNA (mtDNA). MtDNA is almost always only passed to the next generation by females, but under highly exceptional circumstances mtDNA can be passed through males.[clarification needed] The non-recombinant portion of the Y chromosome and the mtDNA, under normal circumstances, do not undergo productive recombination. Part of the Y chromosome can undergo recombination with the X chromosome and within ape history the boundary has changed. Such recombinant changes in the non-recombinant region of Y are extremely rare.[citation needed] Mitochondrial DNA  Illustration of the human mitochondrial DNA with the control region (CR, in grey) containing hypervariable sequences I and II. Mitochondrial DNA became an area of research in phylogenetics in the late 1970s. Unlike genomic DNA, it offered advantages in that it did not undergo recombination. The process of recombination, if frequent enough, corrupts the ability to create parsimonious trees because of stretches of amino acid subsititions (SNPs).[clarification needed] When looking between distantly related species, recombination is less of a problem since recombination between branches from common ancestors is prevented after true speciation occurs. When examining closely related species, or branching within species, recombination creates a large number of 'irrelevant SNPs' for cladistic analysis. MtDNA, through the process of organelle division, became clonal over time; very little, or often none, of that paternal mtDNA is passed. While recombination may occur in mtDNA, there is little risk that it will be passed to the next generation. As a result, mtDNA become clonal copies of each other, except when a new mutation arises. As a result, mtDNA does not have pitfalls of autosomal loci when studied in interbreeding groups. Another advantage of mtDNA is that the hyper-variable regions evolve very quickly; this shows that certain regions of mitochondrial DNA approach neutrality. This allowed the use of mitochondrial DNA to determine that the relative age of the human population was small, having gone through a recent constriction at about 150,000 years ago (see #Causes of errors). Mitochondrial DNA has also been used to verify the proximity of chimpanzees to humans relative to gorillas, and to verify the relationship of these three species relative to the orangutans. 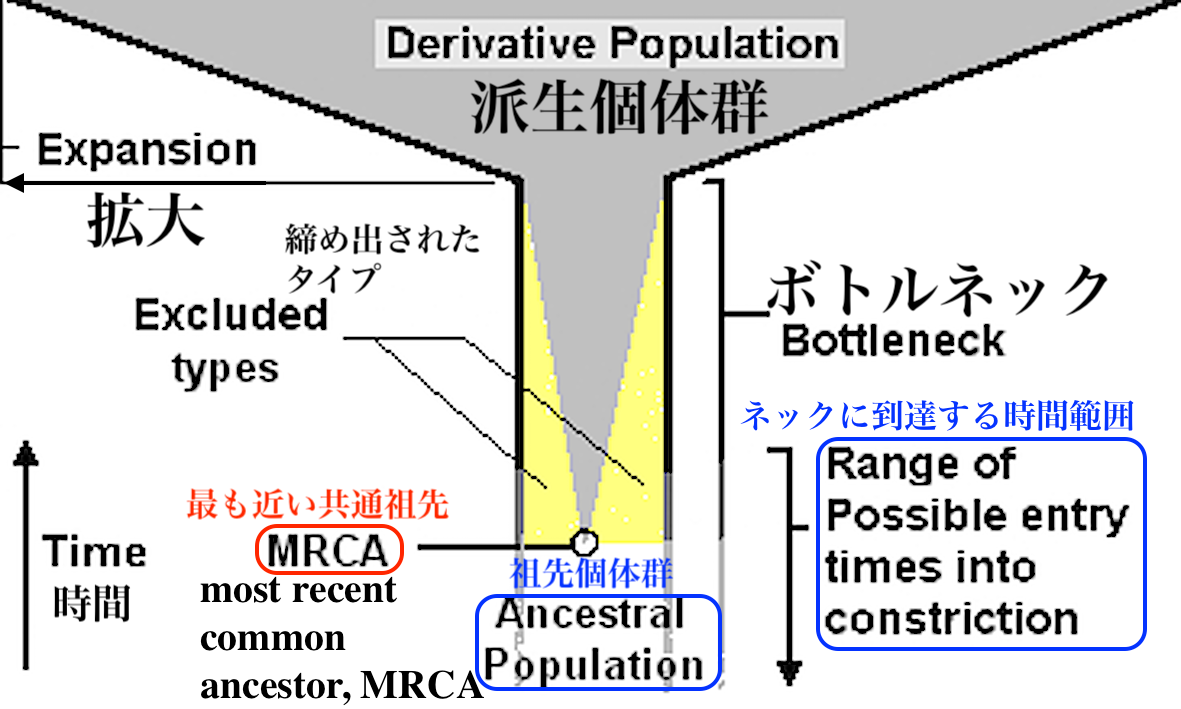 A population bottleneck, as illustrated was detected by intrahuman mtDNA phylogenetic studies; the length of the bottleneck itself is indeterminate per mtDNA. More recently,[when?] the mtDNA genome has been used to estimate branching patterns in peoples around the world, such as when the new world was settled and how. The problem with these studies have been that they rely heavily on mutations in the coding region. Researchers have increasingly discovered that as humans moved from Africa's south-eastern regions, that more mutations accumulated in the coding region than expected, and in passage to the new world some groups are believed[citation needed] to have passed from the Asian tropics to Siberia to an ancient land region called Beringia and quickly migrated to South America. Many of the mtDNA have far more mutations and at rarely mutated coding sites relative to expectations of neutral mutations. Mitochondrial DNA offers another advantage over autosomal DNA. There are generally 2 to 4 copies of each chromosome in each cell (1 to 2 from each parent chromosome). For mtDNA there can be dozens to hundreds in each cell. This increases the amount of each mtDNA loci by at least a magnitude. For ancient DNA, in which the DNA is highly degraded, the number of copies of DNA is helpful in extending and bridging short fragments together, and decreases the amount of bone extracted from highly valuable fossil/ancient remains. Unlike Y chromosome, both male and female remains carry mtDNA in roughly equal quantities.  Schematic of typical animal cell, showing subcellular components. Organelles: (1) nucleolus (2) nucleus (9) mitochondria Y chromosome  Illustration of human Y chromosome The Y chromosome is found in the nucleus of normal cells (nuclear DNA). Unlike mtDNA, it has mutations in the non-recombinant portion (NRY) of the chromosome spaced widely apart, so far apart that finding the mutations on new Y chromosomes is labor-intensive compared with mtDNA. Many studies rely on tandem repeats; however, tandem repeats can expand and retract rapidly and in some predictable patterns. The Y chromosome only tracks male lines, and is not found in females, whereas mtDNA can be traced in males even though they fail to pass on mtDNA. In addition, it has been estimated that effective male populations in the prehistoric period were typically two females per male, and recent studies show that cultural hegemony plays a large role in the passage of Y. This has created discordance between males and females for the Time to the Most Recent Common Ancestor (TMRCA). The estimates for Y TMRCA range from 1/4 to less than 1/2 that of mtDNA TMRCA. It is unclear whether this is due to high male-to-female ratios in the past coupled with repeat migrations from Africa, as a result of mutational rate change, or as some have even proposed that females of the LCA between chimps and humans continued to pass DNA millions after males ceased to pass DNA. At present the best evidence suggests that in migration the male to female ratio in humans may have declined, causing a trimming of Y diversity on multiple occasions within and outside of Africa.  Diagram of human X chromosome showing genetic map For short-range molecular phylogenetics and molecular clocking, the Y chromosome is highly effective and creates a second perspective. One argument that arose was that the Maori by mtDNA appear to have migrated from Eastern China or Taiwan, by Y chromosome from the Papua New Guinea region. When HLA haplotypes were used to evaluate the two hypotheses, it was uncovered that both were right, that the Maori were an admixed population. Such admixtures appear to be common in the human population and thus the use of a single haploid loci can give a biased perspective. |
分子人類学におけるハプロイド遺伝子座 ミトコンドリアの画像。細胞内には多数のミトコンドリアが存在し、その中のDNAは核内の染色体とは独立して複製される。 ヒトには2つの連続した連鎖群があり、1つの性別が持っている。ひとつはY染色体で、父親から息子へと受け継がれる。解剖学的に女性がY染色体を持つの は、遺伝的欠陥の結果として稀である。もう一つの連鎖群はミトコンドリアDNA(mtDNA)である。MtDNAは、ほとんどの場合、女性によってのみ次 世代に受け継がれるが、極めて例外的な状況下では、男性を通じて受け継がれることもある。Y染色体の一部はX染色体と組換えを起こすことがあり、類人猿の 歴史の中でその境界は変化してきた。Yの非組換え領域でこのような組換え変化が起こることは極めて稀である[要出典]。 ミトコンドリアDNA 超可変配列IとIIを含むコントロール領域(CR、灰色)を持つヒトのミトコンドリアDNAの図。 ミトコンドリアDNAは1970年代後半に系統学の研究分野となった。ゲノムDNAとは異なり、組換えを起こさないという利点があった。組み換えが頻繁に 起こると、アミノ酸のサブシテション(SNPs)が伸びるため、簡略化された系統樹を作成する能力が損なわれる[clarification needed]。遠縁の種同士を調べる場合、真の種分化が起こった後に共通の祖先からの枝の間で組み換えが起こらないため、組み換えはあまり問題にならな い。近縁種を調べる場合、あるいは種内の分岐を調べる場合、組換えは系統解析に大量の「無関係なSNP」を作り出す。MtDNAは小器官分裂の過程でク ローン化し、父方のmtDNAはほとんど、あるいは全く受け継がれないことが多い。mtDNAに組換えが起こることはあっても、それが次の世代に受け継が れる危険性はほとんどない。その結果、mtDNAは新たな突然変異が生じた場合を除き、互いにクローン性のコピーとなる。その結果、mtDNAは異種交配 グループで研究する場合、常染色体遺伝子座のような落とし穴がない。mtDNAのもう一つの利点は、超可変領域の進化が非常に速いことである。このこと は、ミトコンドリアDNAのある領域が中立に近づいていることを示している。このことは、ミトコンドリアDNAのある領域が中立に近づいていることを示し ている。このため、ミトコンドリアDNAを使って、人類集団の相対的な年齢が小さく、約15万年前に最近の収縮を経ていることを突き止めることができた (#誤りの原因参照)。 ミトコンドリアDNAはまた、ゴリラに対するチンパンジーの近縁性や、オランウータンに対するこれら3種の近縁性の検証にも使われている。 ヒト内のmtDNA系統学的研究によって、図のような集団ボトルネックが検出された。ボトルネックの長さ自体はmtDNAごとに不確定である。 より最近では、[いつ?]mtDNAゲノムは、新大陸がいつどのように定住したかというような、世界中の民族の分岐パターンを推定するのに使われている。 これらの研究の問題点は、コード領域の突然変異に大きく依存していることである。研究者たちは、人類がアフリカの南東地域から移動するにつれて、コード領 域に予想以上の突然変異が蓄積していることを発見するようになり、新大陸への通過において、アジアの熱帯地域からシベリア、ベリンギアと呼ばれる古代の陸 地地域へと通過し、すぐに南米へと移動したグループもあると考えられている[要出典]。ミトコンドリアDNAの多くは、中立的な変異の予想に比べてはるか に多くの変異を持ち、ほとんど変異のないコード部位に変異がある。 ミトコンドリアDNAには、常染色体DNAにはないもう一つの利点がある。一般的に、各細胞には各染色体のコピーが2〜4本(各親染色体から1〜2本)存 在する。ミトコンドリアDNAの場合、各細胞に数十から数百ある。そのため、mtDNAの各遺伝子座の量は少なくとも1桁は増える。DNAが非常に劣化し ている古代のDNAの場合、DNAのコピー数は、短い断片を延長してつなぎ合わせるのに役立ち、非常に貴重な化石/古代の遺体から抽出される骨の量を減少 させる。Y染色体とは異なり、mtDNAはオスもメスもほぼ同量持っている。 典型的な動物細胞の模式図。細胞小器官:(1)核小体 (2)核 (9)ミトコンドリア Y染色体 ヒトのY染色体の図 Y染色体は正常細胞の核内に存在する(核DNA)。mtDNAとは異なり、染色体の非組換え部分(NRY)に変異があり、その間隔が広いため、新しいY染 色体の変異を見つけるのはmtDNAに比べて手間がかかる。多くの研究はタンデムリピートに依存しているが、タンデムリピートは急速に拡大・縮小する可能 性があり、予測可能なパターンもある。Y染色体はオスのみを追跡し、メスには見られないが、mtDNAはオスがmtDNAを受け継がなかったとしても追跡 することができる。さらに、先史時代の有効な男性集団は、通常男性1人につき女性2人であったと推定されており、最近の研究では、文化的覇権がYの継承に 大きな役割を果たしていることが示されている。YのTMRCAの推定値はmtDNAのTMRCAの1/4から1/2以下である。これが、過去にアフリカか ら繰り返し移住してきた男女比の高さによるものなのか、突然変異率の変化によるものなのか、あるいはチンパンジーとヒトの間のLCAのメスは、オスが DNAを受け継がなくなった後も数百万単位でDNAを受け継いでいたという説もあるように、不明である。現在のところ、最も有力な証拠は、移動の過程でヒ トの男女比が低下し、アフリカ内外で何度もYの多様性が切り捨てられた可能性を示唆している。 ヒトのX染色体の遺伝子地図 短距離分子系統学と分子時計学にとって、Y染色体は非常に有効であり、第二の視点を作り出す。マオリ族はmtDNAでは中国東部か台湾から、Y染色体では パプアニューギニア地域から移動してきたと思われる、という議論が起こった。HLAハプロタイプを使って2つの仮説を評価したところ、どちらも正しく、マ オリ族は混血集団であることが判明した。このような混血はヒトの集団ではよく見られることであり、したがって単一のハプロイド遺伝子座を使用することは 偏った視点を与えることになる。 |
| X-linked studies The X-chromosome is also a form of nuclear DNA. Since it is found as 1 copy in males and 2 non-identical chromosomes in females it has a ploidy of 1.5. However, in humans the effective ploidy is somewhat higher, ~1.7, as females in the breeding population have tended to outnumber males by 2:1 during a large portion of human prehistory. Like mtDNA, X-linked DNA tends to over emphasize female population history much more than male. There have been several studies of loci on X chromosome, in total 20 sites have been examined. These include PDHA1, PDHA1, Xq21.3, Xq13.3, Zfx, Fix, Il2rg, Plp, Gk, Ids, Alas2, Rrm2p4, AmeIX, Tnfsf5, Licam, and Msn. The time to most recent common ancestor (TMRCA) ranges from fixed to ~1.8 million years, with a median around 700ky. These studies roughly plot to the expected fixation distribution of alleles, given linkage disequilibrium between adjacent sites. For some alleles the point of origin is elusive, for others, the point of origin points toward Sub-Saharan Africa. There are some distinctions within SSA that suggest a smaller region, but there is not adequate enough sample size and coverage to define a place of most recent common ancestor. The TMRCA is consistent with and extends the bottleneck implied by mtDNA, confidently to about 500,000 years. |
X連鎖研究 X染色体も核DNAの一種である。男性では1本、女性では2本の非同一染色体が存在するため、倍数性は1.5である。しかし、人類の先史時代の大部分にお いて、繁殖集団における女性の数が男性を2:1で上回る傾向があったため、ヒトの場合、実質的な倍数性はやや高く、〜1.7である。mtDNAと同様に、 X連鎖DNAは男性よりも女性の集団史を過度に強調する傾向がある。X染色体上の遺伝子座についてはいくつかの研究があり、全部で20の部位が調べられて いる。PDHA1、PDHA1、Xq21.3、Xq13.3、Zfx、Fix、Il2rg、Plp、Gk、Ids、Alas2、Rrm2p4、 AmeIX、Tnfsf5、Licam、Msnなどである。最も最近の共通祖先までの時間(TMRCA)は固定年から〜180万年で、中央値は700ky 前後である。これらの研究は、隣接部位間の連鎖不平衡を考慮すると、対立遺伝子の予想固定化分布にほぼ一致する。対立遺伝子の中には起源がはっきりしない ものもあれば、起源がサハラ以南のアフリカを指しているものもある。サブサハラ・アフリカ地域内には、より狭い地域を示唆するいくつかの区別があるが、最 近の共通祖先の場所を定義するには十分なサンプルサイズとカバー率がない。TMRCAはmtDNAが示唆するボトルネックと一致し、その延長線上にある。 |
Autosomal loci Diagram of human karyotype. It shows 22 homologous autosomal chromosome pairs, both the female (XX) and male (XY) versions of the two sex chromosomes, as well as the mitochondrial genome (at bottom left). Further information: Karyotype Rate variation [icon] This section is empty. You can help by adding to it. (July 2010) Ancient DNA sequencing Krings Neandertal mtDNA have been sequenced, and sequence similarity indicates an equally recent origin from a small population on the Neanderthal branch of late hominids. The MCR1 gene has also been sequenced but the results are controversial, with one study claiming that contamination issues cannot be resolved from human Neandertal similarities. Critically, however, no DNA sequence has been obtained from Homo erectus, Homo floresiensis, or any of the other late hominids. Some of the ancient sequences obtained have highly probable errors, and proper control to avoid contamination.  Comparison of differences between human and Neanderthal mtDNA Causes of errors The molecular phylogenetics is based on quantification substitutions and then comparing sequence with other species, there are several points in the process which create errors. The first and greatest challenge is finding "anchors" that allow the research to calibrate the system. In this example, there are 10 mutations between chimpanzee and humans, but the researcher has no known fossils that are agreeably ancestral to both but not ancestral to the next species in the tree, gorilla. However, there are fossils believed to be ancestral to orangutans and humans, from about 14 million years ago. So that the researcher can use orangutan and human comparison and comes up with a difference of 24. Using this he can estimate (24/(14*2, the "2" is for the length of the branch to human (14my) and the branch to orangutan (14 my) from their last common ancestor (LCA). The mutation rate at 0.857 for a stretch of sequence. Mutation rates are given, however, as rate per nucleotide(nt)-site, so if the sequence were say 100 nt in length that rate would be 0.00857/nt per million years. Ten mutations*100nt/(0.00857*2) = 5.8 million years. Problem of calibration There are several problems not seen in the above. First, mutations occur as random events. Second, the chance that any site in the genome varies is different from the next site, a very good example is the codons for amino acids, the first two nt in a codon may mutate at 1 per billion years, but the third nt may mutate 1 per million years. Unless scientist study the sequence of a great many animals, particularly those close to the branch being examined, they generally do not know what the rate of mutation for a given site. Mutations do occur at 1st and 2nd positions of codons, but in most cases these mutations are under negative selection and so are removed from the population over small periods of time. In defining the rate of evolution in the anchor one has the problem that random mutation creates. For example, a rate of .005 or .010 can also explain 24 mutations according to the binomial probability distribution. Some of the mutations that did occur between the two have reverted, hiding an initially higher rate. Selection may play into this, a rare mutation may be selective at point X in time, but later climate may change or the species migrates and it is not longer selective, and pressure exerted on new mutations that revert the change, and sometimes the reversion of a nt can occur, the greater the distance between two species the more likely this is going to occur. In addition, from that ancestral species both species may randomly mutate a site to the same nucleotide. Many times this can be resolved by obtaining DNA samples from species in the branches, creating a parsimonious tree in which the order of mutation can be deduced, creating branch-length diagram. This diagram will then produce a more accurate estimate of mutations between two species. Statistically one can assign variance based on the problem of randomnicity, back mutations, and parallel mutations (homoplasies) in creating an error range. There is another problem in calibration however that has defied statistical analysis. There is a true/false designation of a fossil to a least common ancestor. In reality the odds of having the least common ancestor of two extant species as an anchor is low, often that fossil already lies in one branch (underestimating the age), lies in a third branch (underestimating the age) or in the case of being within the LCA species, may have been millions of years older than the branch. To date the only way to assess this variance is to apply molecular phylogenetics on species claimed to be branch points. This only, however identifies the 'outlying' anchor points. And since it is more likely the more abundant fossils are younger than the branch point the outlying fossil may simply be a rare older representative. These unknowns create uncertainty that is difficult to quantify, and often not attempted. Recent papers have been able to estimate, roughly, variance. The general trend as new fossils are discovered, is that the older fossils underestimated the age of the branch point. In addition to this dating of fossils has had a history of errors and there have been many revised datings. The age assigned by researchers to some major branch points have almost doubled in age over the last 30 years. An excellent example of this is the debate over LM3 (Mungo lake 3) in Australia. Originally it was dated to around 30 ky by carbon dating, carbon dating has problems, however, for sampled over 20ky in age, and severe problems for samples around 30ky in age. Another study looked at the fossil and estimated the age to be 62 ky in age. At the point one has an estimation of mutation rate, given the above there must be two sources of variance that need to be cross-multiplied to generate an overall variance. This is infrequently done in the literature. Problems in estimating TMRCA Time to most recent common ancestor (TMRCA) combines the errors in calibration with errors in determining the age of a local branch. |
常染色体の遺伝子座 ヒトの核型の図。22対の相同な常染色体、2本の性染色体の女性版(XX)と男性版(XY)、そしてミトコンドリアゲノム(左下)が示されている。 さらに詳しい情報はこちら: 核型 変化率 [アイコン] このセクションは空である。追加することで手助けができる。(2010年7月) 古代のDNA配列決定 クリングス・ネアンデルタール人のmtDNAの塩基配列が決定され、配列の類似性から、後期ヒト科のネアンデルタール人枝の小さな集団に由来する、同じく 最近のものであることが示された。MCR1遺伝子の塩基配列も決定されているが、その結果には賛否両論があり、ある研究ではヒトのネアンデルタール人との 類似性からコンタミネーションの問題は解決できないと主張している。しかし決定的に重要なのは、ホモ・エレクトス、ホモ・フローレシエンシス、その他の後 期ヒト科動物のDNA配列は得られていないということである。入手された古代の塩基配列の中には、誤りがある可能性が高いものもあり、汚染を避けるための 適切な管理が必要である。 ヒトとネアンデルタール人のmtDNAの違いの比較 エラーの原因 分子系統学は、置換を定量化し、他の種と塩基配列を比較することに基づいているが、その過程で誤差を生むポイントがいくつかある。最初の、そして最大の難 関は、研究によってシステムを較正するための「アンカー」を見つけることである。この例では、チンパンジーとヒトの間には10の突然変異があるが、研究者 は両者の祖先であることに同意できるが、ツリーの次の種であるゴリラの祖先ではない既知の化石を持っていない。しかし、約1400万年前のオランウータン とヒトの祖先と思われる化石はある。そこで研究者は、オランウータンとヒトを比較し、その差を24とする。これを用いて、彼は最後の共通祖先(LCA)か らヒトへの枝(14my)とオランウータンへの枝(14my)の長さを推定することができる。突然変異率は0.857である。突然変異率はヌクレオチド (nt)部位あたりの突然変異率として与えられるので、仮に配列の長さが100ntだとすると、突然変異率は100万年あたり0.00857/ntとな る。10回の突然変異*100nt/(0.00857*2)=580万年である。 キャリブレーションの問題 上記にはない問題がいくつかある。第一に、突然変異はランダムに起こる。第二に、ゲノムのどの部位が変異しても、次の部位とは異なる確率がある。非常に良 い例がアミノ酸のコドンであり、コドンの最初の2ntは10億年に1回変異するかもしれないが、3nt目は100万年に1回変異するかもしれない。科学者 が多くの動物、特に調査対象の枝に近い動物の塩基配列を調査しない限り、ある部位の突然変異の割合はわからないのが普通である。突然変異はコドンの1位と 2位で起こるが、ほとんどの場合、これらの突然変異は負の選択下にあるため、わずかな期間で集団から取り除かれる。アンカーの進化速度を定義する際、ラン ダム突然変異が引き起こす問題がある。例えば、0.005や0.010の突然変異率でも、二項確率分布に従って24の突然変異を説明することができる。こ の2つの間に起こった突然変異の一部は元に戻り、当初より高い突然変異率が隠されている。希少な突然変異はある時点Xでは選択的であったが、後に気候が変 化したり、種が移動したりして選択的でなくなり、新しい突然変異に圧力がかかり、その突然変異が元に戻ったりするのである。さらに、その祖先種から両方の 種がランダムに同じヌクレオチドに変異する可能性もある。多くの場合、枝分かれしている種からDNAサンプルを入手し、突然変異の順序が推測できるような 簡略化した木を作成し、枝の長さ図を作成することで解決できる。この図は2つの種間の突然変異をより正確に推定する。統計学的には、ランダム性、バック ミューテーション、パラレルミューテーション(ホモプラス)の問題に基づいて分散を割り当て、誤差の範囲を作ることができる。 しかし較正にはもう一つ、統計学的分析が及ばない問題がある。ある化石を最小公倍数祖先とするかしないかという問題である。実際には、2つの現存種の最小 公倍数祖先がアンカーとなる確率は低く、その化石はすでに1つの枝にあったり(年代を過小評価)、第3の枝にあったり(年代を過小評価)、LCA種に含ま れる場合はその枝よりも数百万年古かったりすることが多い。現在までのところ、この分散を評価する唯一の方法は、分岐点と主張される種に分子系統学を適用 することである。しかし、これは「外れた」アンカーポイントを特定するだけである。そして、より豊富な化石は分岐点よりも若い可能性が高いので、外れた化 石は単に稀な古い代表種かもしれない。このような未知の要素は、定量化が難しく、しばしば試みられない不確実性を生み出す。 最近の論文では、おおよその分散を見積もることができるようになった。新しい化石が発見されるにつれ、一般的な傾向として、古い化石は分岐点の年代を過小 評価している。これに加えて、化石の年代測定には誤りがあり、多くの修正年代測定が行われてきた歴史がある。いくつかの主要な分岐点に研究者たちが割り当 てた年代は、過去30年間でほぼ倍増している。その好例が、オーストラリアのLM3(マンゴ湖3)をめぐる論争である。しかし、炭素年代測定法では、 20Ky以上のサンプルには問題があり、30Ky前後のサンプルには深刻な問題がある。別の研究では、この化石の年代を62紀と推定している。 突然変異率を推定する時点では、上記のように2つの分散源があり、それらを掛け合わせて全体的な分散を生成する必要がある。これは文献ではあまり行われていない。 TMRCAの推定における問題点 最近共通祖先までの時間(TMRCA)は較正の誤差とローカルブランチの年齢を決定する誤差を組み合わせたものである。 |
| History Protein era  Structure of human hemoglobin. Hemoglobins from dozens of animals and even plants were sequenced in the 1960s and early 1970s With DNA newly discovered as the genetic material, in the early 1960s protein sequencing was beginning to take off.[4] Protein sequencing began on cytochrome C and Hemoglobin. Gerhard Braunitzer sequenced hemoglobin and myoglobin, in total more than hundreds of sequences from wide-ranging species were done. In 1967 A.C. Wilson began to promote the idea of a "molecular clock". By 1969 molecular clocking was applied to anthropoid evolution and V. Sarich and A.C. Wilson found that albumin and hemoglobin has comparable rates of evolution, indicating chimps and humans split about 4 to 5 million years ago.[5] In 1970, Louis Leakey confronted this conclusion with arguing for improper calibration of molecular clocks.[6] By 1975 protein sequencing and comparative serology combined were used to propose that humans closest living relative (as a species) was the chimpanzee.[7] In hindsight, the last common ancestor (LCA) from humans and chimps appears to older than the Sarich and Wilson estimate, but not as old as Leakey claimed, either. However, Leakey was correct in the divergence of old and new world monkeys, the value Sarich and wilson used was a significant underestimate. This error in prediction capability highlights a common theme. (See Causes of Error) DNA era 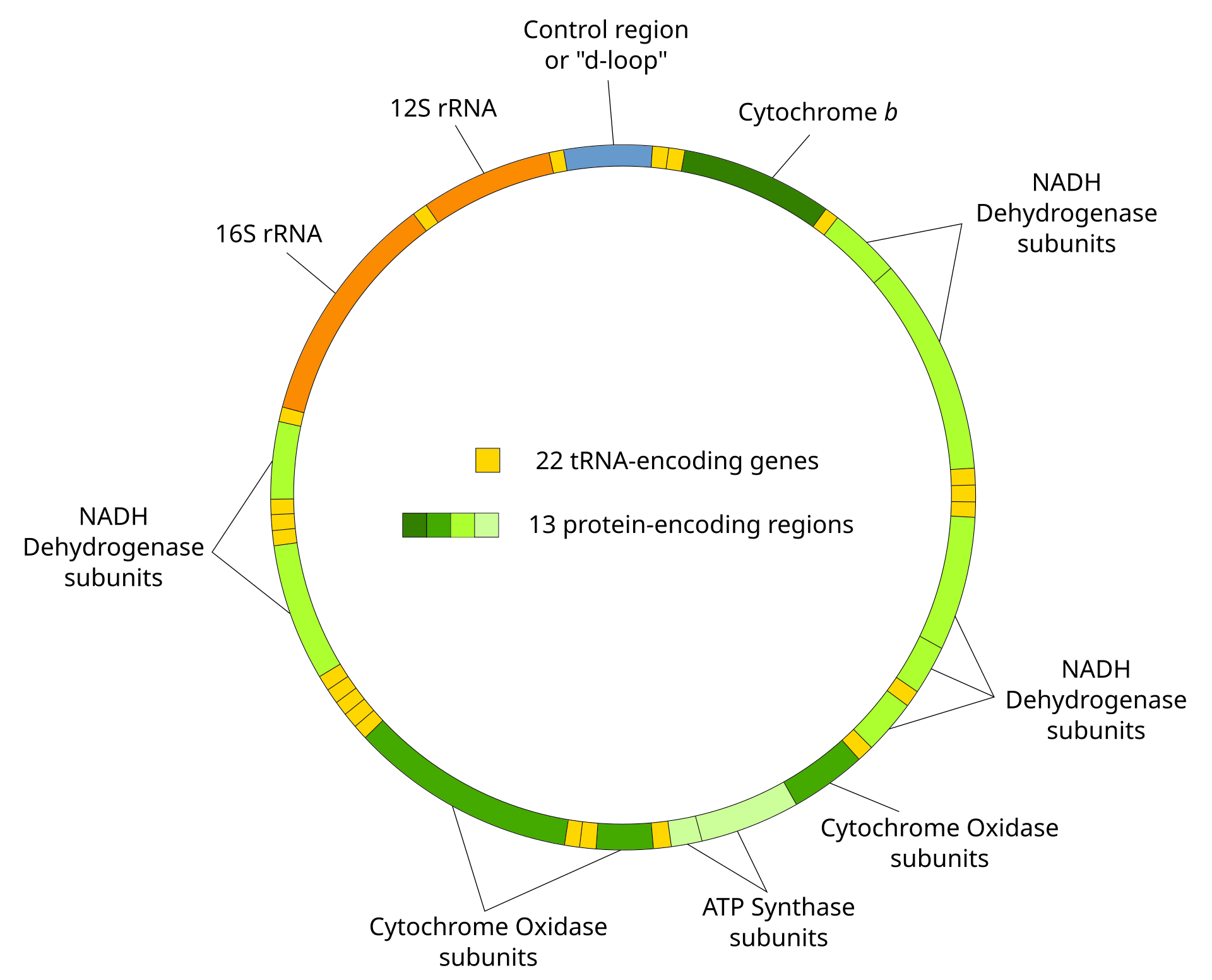 Restriction fragment length polymorphisms studies the cutting of mtDNA into fragments, Later the focus of PCR would be on the D 'control'-loop, at the top of the circle RLFP and DNA hybridization In 1979, W.M.Brown and Wilson began looking at the evolution of mitochondrial DNA in animals, and found they were evolving rapidly.[8] The technique they used was restriction fragment length polymorphism (RFLP), which was more affordable at the time compared to sequencing. In 1980, W.M. Brown, looking at the relative variation between human and other species, recognized there was a recent constriction (180,000 years ago) in the human population.[9] A year later Brown and Wilson were looking at RFLP fragments and determined the human population expanded more recently than other ape populations.[10] In 1984 the first DNA sequence from an extinct animal was done.[11] Sibley and Ahlquist apply DNA-DNA hybridization technology to anthropoid phylogeny, and see pan/human split closer than gorilla/pan or gorilla/human split, a highly controversial claim.[12][13] However, in 1987 they were able to support their claim.[14] In 1987, Cann, Stoneking and Wilson suggest, by RFLP analysis of human mitochondrial DNA, that humans evolved from a constrict in Africa of a single female in a small population, ~10,00 individuals, 200,000 years ago.[15] Era of PCR 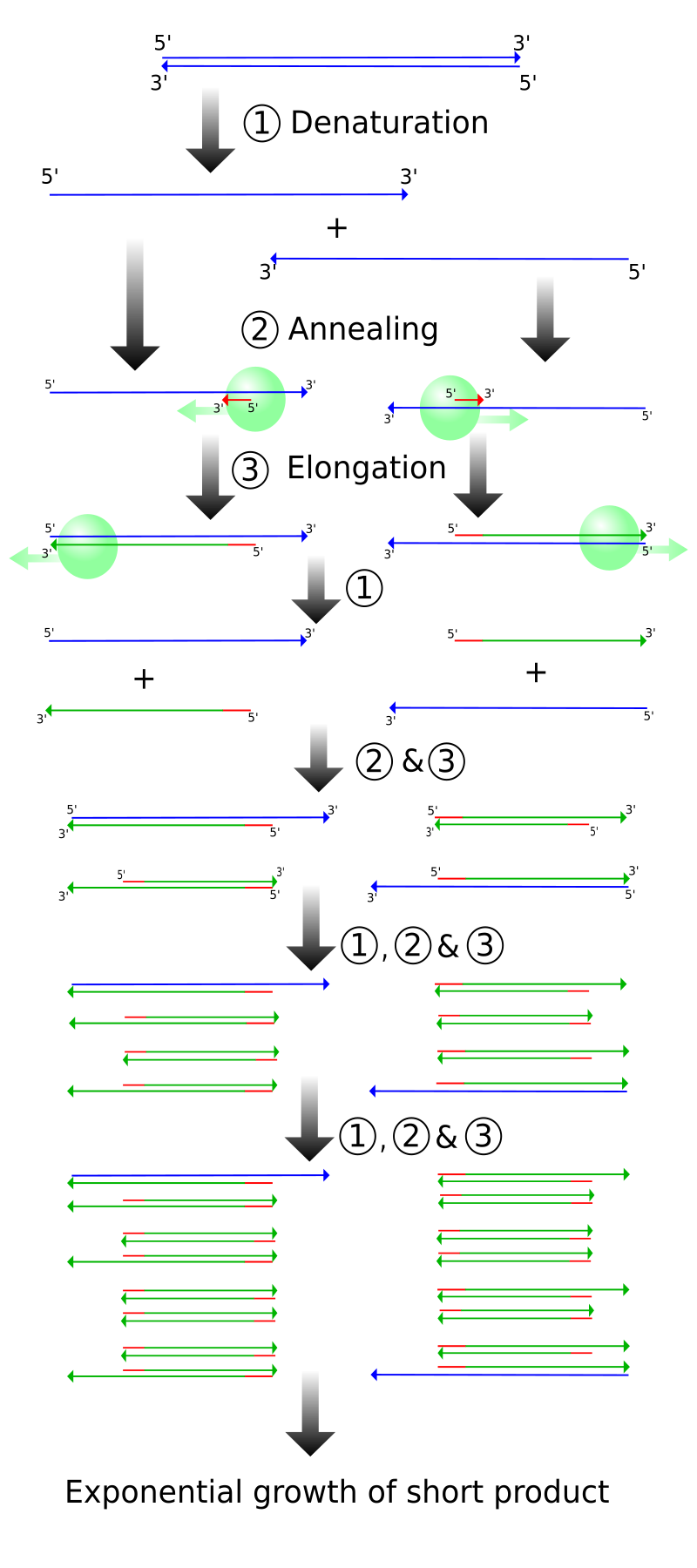 PCR could rapidly amplify DNA from one molecule to billions, allowing sequencing from human hairs or ancient DNA. In 1987, PCR-amplification of mtDNA was first used to determine sequences.[16] In 1991 Vigilante et al. published the seminal work on mtDNA phylogeny implicating sub-saharan Africa as the place of humans most recent common ancestors for all mtDNAs.[17] The war between out-of-Africa and multiregionalism, already simmering with the critiques of Allan Templeton, soon escalated with the paleoanthropologist, like Milford Wolpoff, getting involved.[18][19][20] In 1995, F. Ayala published his critical Science article "The Myth about Eve", which relied on HLA-DR sequence.[21] At the time, however Ayala was not aware of rapid evolution of HLA loci via recombinatory process. In 1996, Parham and Ohta published their finds on the rapid evolution of HLA by short-distance recombination ('gene conversion' or 'abortive recombination'), weakening Ayala's claim (Parham had actually written a review a year earlier, but this had gone unnoticed).[22][23] A stream of papers would follow from both sides, many with highly flawed methods and sampling. One of the more interesting[according to whom?] was Harris and Hey, 1998 which showed that the TMCRA (time to most recent common ancestor) for the PDHA1 gene was well in excess of 1 million years. Given a ploidy at this locus of 1.5 (3 fold higher than mtDNA) the TMRCA was more than double the expectation. While this falls into the 'fixation curve' of 1.5 ploidy (averaging 2 female and 1 male) the suggested age of 1.8 my is close a significantly deviant p-value for the population size, possibly indicating that the human population shrank or split off of another population.[24] Oddly, the next X-linked loci they examined, Factor IX, showed a TMRCA of less than 300,000 years.[25]  Cross-linked DNA extracted from the 4,000-year-old liver of an Ancient Egyptian priest called Nekht-Ankh Ancient DNA Ancient DNA sequencing had been conducted on a limited scale up to the late 1990s when the staff at the Max Planck Institute shocked the anthropology world by sequencing DNA from an estimated 40,000-year-old Neanderthal.[26][27][28] The result of that experiment is that the differences between humans living in Europe, many of which were derived from haplogroup H (CRS), Neandertals branched from humans more than 300,000 years before haplogroup H reached Europe. While the mtDNA and other studies continued to support a unique recent African origin, this new study basically answered critiques from the Neandertal side. Genomic sequencing Significant progress has been made in genomic sequencing since Ingman and colleague published their finding on mitochondrial genome.[29] Several papers on genomic mtDNA have been published; there is considerable variability in the rate of evolution, and rate variation and selection are evident at many sites. In 2007, Gonder et al. proposed that a core population of humans, with greatest level of diversity and lowest selection, once lived in the region of Tanzania and proximal parts of southern Africa, since humans left this part of Africa, mitochondria have been selectively evolving to new regions.[30] Critical progress Critical in the history of molecular anthropology: That molecular phylogenetics could compete with comparative anthropology for determining the proximity of species to humans. Wilson and King realized in 1975, that while there was equity between the level of molecular evolution branching from chimp to human to putative LCA, that there was an inequity in morphological evolution. Comparative morphology based on fossils could be biased by different rates of change.[7] Realization that in DNA there are multiple independent comparisons. Two techniques, mtDNA and hybridization converge on a single answer, chimps as a species are most closely related to humans. The ability to resolve population sizes based on the 2N rule, proposed by Kimura in the 1950s.[31] To use that information to compare relative sizes of population and come to a conclusion about abundance that contrasted observations based on the paleontological record. While human fossils in the early and middle stone age are far more abundant than chimpanzee or gorilla, there are few unambiguous chimpanzee or gorilla fossils from the same period Loci that have been used in molecular phylogenetics: Cytochrome C Serum albumin Hemoglobin - Braunitizer, 1960s, Harding et al. 1997 Mitochondrial D-loop - Wilson group, 1980, 1981, 1984, 1987, 1989, 1991(posthumously) - TMRCA about 170 kya. Y-chromosome HLA-DR - Ayala 1995 - TMRCA for locus is 60 million years. CD4 (Intron) - Tishkoff, 1996 - most of the diversity is in Africa. PDHA1 (X-linked) Harris and Hey - TMRCA for locus greater than 1.5 million years. Xlinked loci: PDHA1, Xq21.3, Xq13.3, Zfx, Fix, Il2rg, Plp, Gk, Ids, Alas2, Rrm2p4, AmeIX, Tnfsf5, Licam, and Msn Autosomal:Numerous. |
歴史 タンパク質の時代 ヒトのヘモグロビンの構造。1960年代から1970年代初頭にかけて、数十種類の動物や植物からもヘモグロビンの塩基配列が決定された。 遺伝物質としてDNAが新たに発見され、1960年代初頭にはタンパク質の配列決定が始まった。ゲルハルト・ブラウニッツァーはヘモグロビンとミオグロビ ンの塩基配列を決定し、幅広い生物種から合計数百以上の塩基配列が決定された。1967年、A.C.ウィルソンは「分子時計」の考えを広め始めた。 1969年までに分子時計は人類進化に応用され、V.サリッチとA.C.ウィルソンはアルブミンとヘモグロビンが同程度の進化速度であることを発見し、チ ンパンジーとヒトが約400万年から500万年前に分裂したことを示した[5]。1970年、ルイス・リーキーは分子時計の不適切な較正を主張し、この結 論と対立した[6]。 [今にして思えば、ヒトとチンパンジーの最後の共通祖先(LCA)は、サリッチとウィルソンの推定よりも古いようだが、リーキーが主張したほど古くもな い。しかし、旧世界猿と新世界猿の分岐においてリーキーは正しく、サリッチとウィルソンが用いた値はかなり過小評価であった。この予測能力の誤差は、共通 のテーマを浮き彫りにしている。(誤差の原因参照) DNA時代 制限断片長多型はmtDNAを断片に切断する研究であり、その後PCRの焦点は円の一番上にあるD「コントロール」ループに当てられるようになった。 RLFPとDNAハイブリダイゼーション 1979年、W.M.ブラウンとウィルソンは動物のミトコンドリアDNAの進化を調べ始め、それらが急速に進化していることを発見した。1980年、 W.M.ブラウンはヒトと他の種との相対的変異を調べ、ヒトの集団に最近(18万年前)の狭窄があることを認識した[9]。 [11]シブリーとアールキストはDNA-DNAハイブリダイゼーション技術を類人猿系統学に応用し、パン/ヒトの分裂はゴリラ/パンやゴリラ/ヒトの分 裂よりも近いと見ており、この主張は非常に議論を呼んだ[12][13]。 [14】1987年、カン、ストーンキングとウィルソンは、ヒトのミトコンドリアDNAのRFLP分析によって、ヒトは20万年前にアフリカの小さな集 団、〜10,00個体、1匹のメスから進化したことを示唆した[15]。 PCRの時代 PCR法は、1分子から数十億分子までのDNAを迅速に増幅することができるため、ヒトの毛髪や古代のDNAから塩基配列を決定することが可能になった。 1987年、mtDNAのPCR増幅が初めて塩基配列の決定に用いられた[16]。1991年、Vigilanteらは、mtDNAの系統学に関する代表 的な研究結果を発表し、サハラ以南のアフリカが、全てのmtDNAの最も最近の共通祖先の場所であることを示唆した[17]。 [17]アラン・テンプルトンの批評ですでに煮えたぎっていたアフリカ外派と多地域派との抗争は、ミルフォード・ウォルポフのような古人類学者を巻き込ん ですぐにエスカレートした[18][19][20]。 1995年、F.アヤラはHLA-DR配列に依拠した批判的なサイエンス論文「イヴについての神話」を発表した[21]。1996年、Parhamと太田 は短距離組換え(「遺伝子転換」または「中止組換え」)によるHLAの急速な進化に関する発見を発表し、アヤラの主張を弱めた(Parhamはその1年前 に実際に総説を書いていたが、これは気づかれなかった)[22][23]。より興味深い論文の1つは[誰によると]、PDHA1遺伝子のTMCRA(最も 最近の共通祖先までの時間)が100万年をはるかに超えていたことを示したHarris and Hey, 1998である。この遺伝子座の倍数が1.5(mtDNAの3倍)であることを考えると、TMRCAは予想の2倍以上であった。これは1.5倍体(平均し て女性2人、男性1人)の「固定曲線」に当てはまるが、1.8万年という示唆された年齢は、集団規模のp値としては著しく逸脱しており、おそらくヒト集団 が縮小したか、別の集団から分裂したことを示している[24]。奇妙なことに、彼らが次に調べたX-連鎖遺伝子座、第IX因子のTMRCAは30万年未満 であった[25]。 Nekht-Ankhと呼ばれる古代エジプトの司祭の4,000年前の肝臓から抽出された架橋DNAは、TMRCAが30万年未満であった[25]。 古代DNA 1990年代後半にマックス・プランク研究所のスタッフが推定4万年前のネアンデルタール人からDNAの塩基配列を決定し、人類学の世界に衝撃を与えた時 までは、古代DNAの塩基配列決定は限定的な規模で行われていた[26][27][28]。その実験の結果、ヨーロッパに住む人類との違いは、その多くが ハプログループH(CRS)に由来するものであり、ネアンデルタール人はハプログループHがヨーロッパに到達する30万年以上前に人類から分岐していたこ とが判明した。mtDNAやその他の研究は、最近のアフリカ起源というユニークな説を支持し続けていたが、この新しい研究は基本的にネアンデルタール人側 からの批判に答えたものである。 ゲノム解読 Ingmanとその同僚がミトコンドリアゲノムに関する発見を発表して以来、ゲノムの塩基配列決定において大きな進展があった[29]。ゲノムの mtDNAに関する論文がいくつか発表されているが、進化の速度にはかなりのばらつきがあり、多くの部位で速度の変化と選択が見られる。2007年、ゴン デールらは、最も多様性のレベルが高く、選択度が最も低いヒトの中核集団が、かつてタンザニアと南部アフリカの近辺に住んでおり、ヒトがアフリカのこの地 域を離れて以来、ミトコンドリアは新しい地域へと選択的に進化してきたと提唱した[30]。 重要な進歩 分子人類学の歴史において重要なことである: 分子系統学は、人類に近い種を決定するための比較人類学と競合することができた。 ウィルソンとキングは1975年、チンプからヒト、そして推定LCAへと分岐する分子進化のレベルには公平性がある一方で、形態学的進化には不公平性があることに気づいた。化石に基づく比較形態学は、異なる変化率によって偏りが生じる可能性があった[7]。 DNAには複数の独立した比較対象が存在することに気づいた。mtDNAとハイブリダイゼーションという2つの手法は、チンパンジーという種がヒトに最も近縁であるという1つの答えに収束する。 1950年代に木村によって提唱された2N法則に基づき、集団の大きさを決定する能力[31]。その情報を使って集団の相対的な大きさを比較し、古生物学 的記録に基づく観察と対照的な豊度に関する結論を導き出した。初期・中期石器時代のヒトの化石は、チンパンジーやゴリラよりもはるかに豊富である一方、同 時代のチンパンジーやゴリラの化石には明確なものはほとんどない。 分子系統学で使用されてきた遺伝子: シトクロムC 血清アルブミン ヘモグロビン - Braunitizer, 1960s, Harding et al. ミトコンドリアDループ - Wilsonグループ、1980年、1981年、1984年、1987年、1989年、1991年(死後) - TMRCA約170kya。 Y染色体 HLA-DR - Ayala 1995年 - 遺伝子座のTMRCAは6000万年である。 CD4(イントロン) - Tishkoff, 1996 - 多様性のほとんどはアフリカにある。 PDHA1(X連鎖) Harris and Hey - 遺伝子座のTMRCAは150万年以上である。 X連鎖遺伝子座: PDHA1、Xq21.3、Xq13.3、Zfx、Fix、Il2rg、Plp、Gk、Ids、Alas2、Rrm2p4、AmeIX、Tnfsf5、Licam、Msn。 常染色体:多数ある。 |
| https://en.wikipedia.org/wiki/Molecular_anthropology |
|
| Most recent common ancestor, MRCA In biology and genetic genealogy, the most recent common ancestor (MRCA), also known as the last common ancestor (LCA), of a set of organisms is the most recent individual from which all the organisms of the set are descended. The term is also used in reference to the ancestry of groups of genes (haplotypes) rather than organisms. The MRCA of a set of individuals can sometimes be determined by referring to an established pedigree. However, in general, it is impossible to identify the exact MRCA of a large set of individuals, but an estimate of the time at which the MRCA lived can often be given. Such time to most recent common ancestor (TMRCA) estimates can be given based on DNA test results and established mutation rates as practiced in genetic genealogy, or by reference to a non-genetic, mathematical model or computer simulation. In organisms using sexual reproduction, the matrilineal MRCA and patrilineal MRCA are the MRCAs of a given population considering only matrilineal and patrilineal descent, respectively. The MRCA of a population by definition cannot be older than either its matrilineal or its patrilineal MRCA. In the case of Homo sapiens, the matrilineal and patrilineal MRCA are also known as "Mitochondrial Eve" (mt-MRCA) and "Y-chromosomal Adam" (Y-MRCA) respectively. The age of the human MRCA is unknown. It is no greater than the age of either the Y-MRCA or the mt-MRCA, estimated at around 200,000 years. Unlike in pedigrees of individual humans or domesticated lineages where historical parentage is known, in the inference of relationships among species or higher groups of taxa (systematics or phylogenetics), ancestors are not directly observable or recognizable. They are inferences based on patterns of relationship among taxa inferred in a phylogenetic analysis of extant organisms and/or fossils.[1] The last universal common ancestor (LUCA) is the most recent common ancestor of all current life on Earth, estimated to have lived some 3.5 to 3.8 billion years ago (in the Paleoarchean).[2][3][note 1] |
もっとも近い共通の祖先(Most recent common ancestor) 生物学や遺伝系譜学では、ある生物の集合の最も最近の共通祖先(MRCA)(最後の共通祖先(LCA)とも呼ばれる)とは、その集合のすべての生物の子孫となる最も最近の個体のことである。この用語は、生物ではなく遺伝子群(ハプロタイプ)の祖先についても使われる。 個体群のMRCAは、確立された血統を参照することで決定できることも ある。しかし一般的に、多数の個体のMRCAを正確に特定することは不可能である。このような最も最近の共通祖先までの時間(TMRCA)の推定は、遺伝 系図学で行われているDNA検査結果と確立された突然変異率に基づいて、あるいは遺伝学的でない数学的モデルやコンピューターシミュレーションを参照して 行うことができる。 有性生殖を用いる生物では、母系MRCAと父系MRCAはそれぞれ母系血統と父系血統のみを考慮したある集団のMRCAである。定義上、ある集団の MRCAは母系MRCAと父系MRCAのどちらよりも古くなることはない。ホモ・サピエンスの場合、母系MRCAと父系MRCAはそれぞれ「ミトコンドリ ア・イブ」(mt-MRCA)と「Y染色体アダム」(Y-MRCA)としても知られている。 ヒトのMRCAの年齢は不明である。Y-MRCAやmt-MRCAの年齢を上回ることはなく、およそ20万年と推定されている。 歴史的な親がわかっている個々のヒトや家畜化された系統の血統とは異なり、種や高次の分類群間の関係を推定する場合(系統学または系統学)、祖先は直接観 察できるものでも認識できるものでもない。現存する生物や化石の系統解析から推測される分類群間の関係パターンに基づく推論である[1]。 最後の普遍的共通祖先(LUCA)とは、地球上の現生命体の中で最も新しい共通祖先であり、約35億年から38億年前(古明代)に生きていたと推定されている[2][3][注釈 1]。 |
| MRCA of different species Further information: Chimpanzee–human last common ancestor, Last universal common ancestor, Phylogenetic tree, and Tree of life (biology) 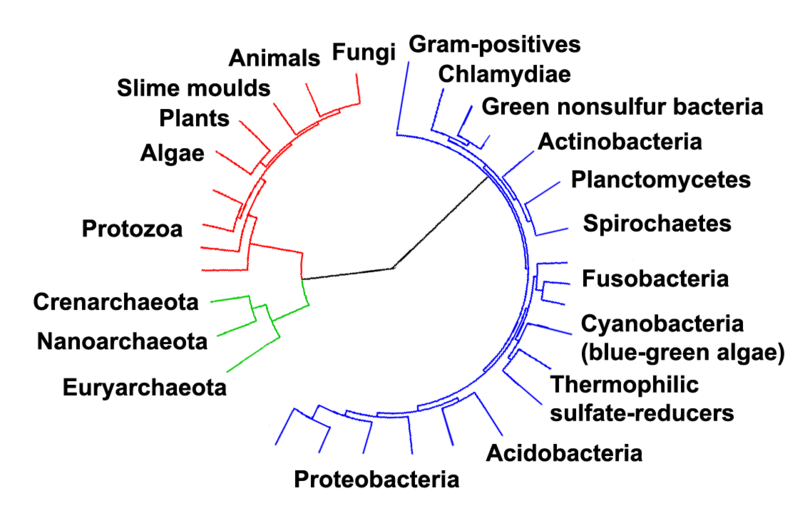 Evolutionary tree showing the divergence of modern species from the last universal ancestor in the center.[5] The three domains are colored, with bacteria blue, archaea green, and eukaryotes red. The project of a complete description of the phylogenetic relationships among all biological species is dubbed the "tree of life". This involves inference of ages of divergence for all hypothesized clades; for example, the MRCA of all Carnivora (i.e. the MRCA of "cats and dogs") is estimated to have diverged some 42 million years ago (Miacidae).[6] The concept of the last common ancestor from the perspective of human evolution is described for a popular audience in The Ancestor's Tale by Richard Dawkins (2004). Dawkins lists "concestors" of the human lineage in order of increasing age, including hominin (human–chimpanzee), hominine (human–gorilla), hominid (human–orangutan), hominoid (human–gibbon), and so on in 40 stages in total, down to the last universal common ancestor (human–bacteria). |
異なる種のMRCA さらに詳しい情報 チンパンジーとヒトの最後の共通祖先、最後の普遍的共通祖先、系統樹、生命の樹(生物学) 中央の最後の普遍的共通祖先からの現生種の分岐を示す進化ツリー[5]。3つのドメインは、バクテリアは青、古細菌は緑、真核生物は赤で色分けされている。 すべての生物種の系統関係を完全に記述するプロジェクトは、「生命の樹」と呼ばれている。例えば、すべての食肉目のMRCA(すなわち「ネコとイヌ」のMRCA)は、約4200万年前に分岐したと推定されている(ミアカ科)[6]。 人類進化の観点から見た最後の共通祖先の概念は、リチャード・ドーキンス著『The Ancestor's Tale』(2004年)に一般向けに記述されている。ドーキンスは、ヒトの系統の「同祖」を年齢の高い順に列挙している。ホミニン(ヒト-チンパン ジー)、ホミニン(ヒト-ゴリラ)、ホミニッド(ヒト-オランウータン)、ホミニッド(ヒト-テナガザル)など、全部で40段階あり、最後の普遍的共通祖 先(ヒト-バクテリア)に至る。 |
| MRCA of a population identified by a single genetic marker Main article: Coalescent theory It is also possible to consider the ancestry of individual genes (or groups of genes, haplotypes) instead of an organism as a whole. Coalescent theory describes a stochastic model of how the ancestry of such genetic markers maps to the history of a population. Unlike organisms, a gene is passed down from a generation of organisms to the next generation either as perfect replicas of itself or as slightly mutated descendant genes. While organisms have ancestry graphs and progeny graphs via sexual reproduction, a gene has a single chain of ancestors and a tree of descendants. An organism produced by sexual cross-fertilization (allogamy) has at least two ancestors (its immediate parents), but a gene always has one ancestor per generation. |
単一の遺伝マーカーによって特定される集団のMRCA 主な記事 コアレセント理論 生物全体ではなく、個々の遺伝子(または遺伝子群、ハプロタイプ)の祖先を考えることも可能である。Coalescent theoryは、このような遺伝マーカーの祖先が集団の歴史にどのようにマッピングされるかを確率論的にモデル化したものである。 生物とは異なり、遺伝子は生物の世代から次の世代へと、それ自身の完全な複製として、あるいはわずかに変異した子孫遺伝子として受け継がれる。生物は有性 生殖によって祖先のグラフと子孫のグラフを持つが、遺伝子は祖先の一本の鎖と子孫の木を持つ。有性交配(アロガミー)によって生まれた生物は、少なくとも 2つの祖先(直系の親)を持つが、遺伝子は1世代につき常に1つの祖先を持つ。 |
| Patrilineal and matrilineal MRCA Main articles: Mitochondrial Eve and Y-chromosomal Adam 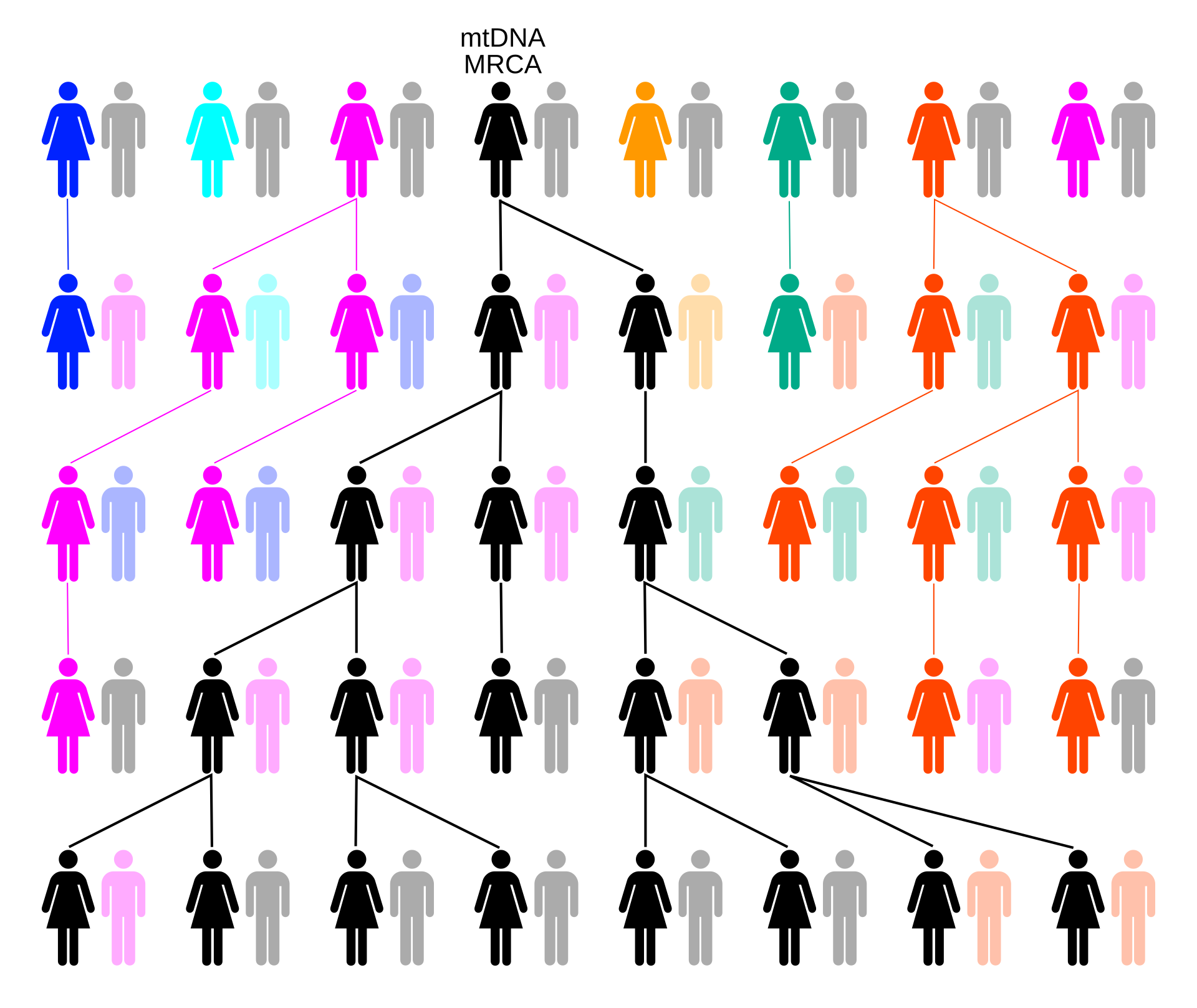 Through random drift or selection, lineage will trace back to a single person. In this example over 5 generations, the colors represent extinct matrilineal lines and black the matrilineal line descended from the mt-MRCA. Mitochondrial DNA (mtDNA) is nearly immune to sexual mixing, unlike the nuclear DNA whose chromosomes are shuffled and recombined in Mendelian inheritance. Mitochondrial DNA, therefore, can be used to trace matrilineal inheritance and to find the Mitochondrial Eve (also known as the African Eve), the most recent common ancestor of all humans via the mitochondrial DNA pathway. Likewise, Y chromosome is present as a single sex chromosome in the male individual and is passed on to male descendants without recombination. It can be used to trace patrilineal inheritance and to find the Y-chromosomal Adam, the most recent common ancestor of all humans via the Y-DNA pathway. Approximate dates for Mitochondrial Eve and Y-chromosomal Adam have been established by researchers using genealogical DNA tests. Mitochondrial Eve is estimated to have lived about 200,000 years ago. A paper published in March 2013 determined that, with 95% confidence and that provided there are no systematic errors in the study's data, Y-chromosomal Adam lived between 237,000 and 581,000 years ago.[7][8] The MRCA of all humans alive today would, therefore, need to have lived more recently than either.[9][note 2] It is more complicated to infer human ancestry via autosomal chromosomes. Although an autosomal chromosome contains genes that are passed down from parents to children via independent assortment from only one of the two parents, genetic recombination (chromosomal crossover) mixes genes from non-sister chromatids from both parents during meiosis, thus changing the genetic composition of the chromosome. |
父系制と母系制のMRCA 主な記事 ミトコンドリア・イブとY染色体アダム ランダムなドリフトや淘汰によって、血統は一人の人間にさかのぼる。この5世代にわたる例では、色は絶滅した母系血統、黒はmt-MRCAから派生した母系血統を表している。 ミトコンドリアDNA(mtDNA)は、メンデル遺伝で染色体がシャッフルされ、組み替えられる核DNAとは異なり、性的な混合の影響をほとんど受けな い。従って、ミトコンドリアDNAは母系遺伝を追跡し、ミトコンドリアDNA経路を通じて全人類の最も新しい共通祖先であるミトコンドリア・イブ(アフリ カン・イヴとも呼ばれる)を見つけるのに使うことができる。 同様に、Y染色体は男性個体に単一の性染色体として存在し、組換えなしに男性の子孫に受け継がれる。父系遺伝をたどり、Y-DNA経路を通じて全人類の最も新しい共通祖先であるY染色体アダムを見つけるために使用できる。 ミトコンドリア・イブとY染色体アダムのおおよその年代は、系図DNA検査を用いた研究者によって確立されている。ミトコンドリア・イブは約20万年前に 生きていたと推定されている。2013年3月に発表された論文では、信頼度95%で、研究データに系統的な誤りがなければ、Y染色体アダムは 237,000~581,000年前に生きていたと断定している[7][8]。 したがって、現在生きている全人類のMRCAは、どちらかよりも最近に生きていた必要がある[9][注 2]。 常染色体からヒトの祖先を推定するのはより複雑である。常染色体には、両親のどちらか一方から独立した遺伝子が受け継がれる。しかし、減数分裂の際に、両親の姉妹染色分体以外の遺伝子が混ざり合い、染色体の遺伝的構成が変化する。 |
| Time to MRCA estimates Different types of MRCAs are estimated to have lived at different times in the past. These time to MRCA (TMRCA) estimates are also computed differently depending on the type of MRCA being considered. Patrilineal and matrilineal MRCAs (Mitochondrial Eve and Y-chromosomal Adam) are traced by single gene markers, thus their TMRCA are computed based on DNA test results and established mutation rates as practiced in genetic genealogy. The time to the genealogical MRCA (most recent common ancestor by any line of descent) of all living humans cannot be traced genetically because the DNA of the great majority of ancestors is completely lost after a few hundred years. It is therefore computed based on non-genetic, mathematical models and computer simulations. Since Mitochondrial Eve and Y-chromosomal Adam are traced by single genes via a single ancestral parent line, the time to these genetic MRCAs will necessarily be greater than that for the genealogical MRCA. This is because single genes will coalesce more slowly than tracing of conventional human genealogy via both parents. The latter considers only individual humans, without taking into account whether any gene from the computed MRCA actually survives in every single person in the current population.[11] TMRCA via genetic markers Mitochondrial DNA can be used to trace the ancestry of a set of populations. In this case, populations are defined by the accumulation of mutations on the mtDNA, and special trees are created for the mutations and the order in which they occurred in each population. The tree is formed through the testing of a large number of individuals all over the world for the presence or lack of a certain set of mutations. Once this is done it is possible to determine how many mutations separate one population from another. The number of mutations, together with estimated mutation rate of the mtDNA in the regions tested, allows scientists to determine the approximate time to MRCA (TMRCA) which indicates time passed since the populations last shared the same set of mutations or belonged to the same haplogroup. In the case of Y-Chromosomal DNA, TMRCA is arrived at in a different way. Y-DNA haplogroups are defined by single-nucleotide polymorphism in various regions of the Y-DNA. The time to MRCA within a haplogroup is defined by the accumulation of mutations in STR sequences of the Y-Chromosome of that haplogroup only. Y-DNA network analysis of Y-STR haplotypes showing a non-star cluster indicates Y-STR variability due to multiple founding individuals. Analysis yielding a star cluster can be regarded as representing a population descended from a single ancestor. In this case the variability of the Y-STR sequence, also called the microsatellite variation, can be regarded as a measure of the time passed since the ancestor founded this particular population. The descendants of Genghis Khan or one of his ancestors represents a famous star cluster that can be dated back to the time of Genghis Khan.[12] TMRCA calculations are considered critical evidence when attempting to determine migration dates of various populations as they spread around the world. For example, if a mutation is deemed to have occurred 30,000 years ago, then this mutation should be found amongst all populations that diverged after this date. If archeological evidence indicates cultural spread and formation of regionally isolated populations then this must be reflected in the isolation of subsequent genetic mutations in this region. If genetic divergence and regional divergence coincide it can be concluded that the observed divergence is due to migration as evidenced by the archaeological record. However, if the date of genetic divergence occurs at a different time than the archaeological record, then scientists will have to look at alternate archaeological evidence to explain the genetic divergence. The issue is best illustrated in the debate surrounding the demic diffusion versus cultural diffusion during the European Neolithic.[13] TMRCA of all living humans The age of the MRCA of all living humans is unknown. It is necessarily younger than the age of either the matrilinear or the patrilinear MRCA, both of which have an estimated age of between roughly 100,000 and 200,000 years ago.[14] A study by mathematicians Joseph T. Chang, Douglas Rohde and Steve Olson used a theoretical model to calculate that the MRCA may have lived remarkably recently, possibly as recently as 2,000 years ago. It concludes that the MRCA of all humans probably lived in East Asia, which would have given them key access to extremely isolated populations in Australia and the Americas. Possible locations for the MRCA include places such as the Chuckchi and Kamchatka Peninsulas that are close to Alaska, places such as Indonesia and Malaysia that are close to Australia or a place such as Taiwan or Japan that is more intermediate to Australia and the Americas. European colonization of the Americas and Australia was found by Chang to be too recent to have had a substantial impact on the age of the MRCA. In fact, if the Americas and Australia had never been discovered by Europeans, the MRCA would only be about 2.3% further back in the past than it is.[15][16][17] Note that the age of the MRCA of a population does not correspond to a population bottleneck, let alone a "first couple". It rather reflects the presence of a single individual with high reproductive success in the past, whose genetic contribution has become pervasive throughout the population over time. It is also incorrect to assume that the MRCA passed all, or indeed any, genetic information to every living person. Through sexual reproduction, an ancestor passes half of his or her genes to each descendant in the next generation; in the absence of pedigree collapse, after just 32 generations the contribution of a single ancestor would be on the order of 2−32, a number proportional to less than a single basepair within the human genome.[18] |
MRCA推定までの時間 MRCAの種類によって、過去に生息していた時期は異なる。これらのMRCAまでの時間(TMRCA)の推定値も、考慮されるMRCAのタイプによって計 算方法が異なる。父系的MRCAと母系的MRCA(ミトコンドリア・イブとY染色体アダム)は単一遺伝子マーカーによって追跡されるため、そのTMRCA はDNA検査結果と遺伝的系譜学で実践されている確立された突然変異率に基づいて計算される。現存する全人類の系図上のMRCA(あらゆる血統による最近 の共通祖先)までの時間は、遺伝学的に追跡することができない。なぜなら、大多数の祖先のDNAは数百年後には完全に失われてしまうからである。そのた め、遺伝学的ではない数学的モデルやコンピューターシミュレーションに基づいて計算される。 ミトコンドリア・イブとY染色体アダムは、単一の祖先の親系統を経由した単一遺伝子によって追跡されるため、これらの遺伝的MRCAまでの時間は、系図的 MRCAの時間よりも必然的に長くなる。というのも、単一遺伝子の合体は、両親を経由する従来のヒトの系譜をたどるよりも遅いからである。後者では、計算 されたMRCAに由来する遺伝子が実際に現在の集団のすべての人の中に生存しているかどうかを考慮することなく、個々の人間のみを考慮する[11]。 遺伝子マーカーによるTMRCA ミトコンドリアDNAは、集団の祖先を追跡するために使用することができる。この場合、集団はmtDNA上の突然変異の蓄積によって定義され、各集団にお ける突然変異とその発生順序について特別な樹が作成される。この樹は、世界中の多数の個体について、特定の突然変異の有無を調べることによって形成され る。一旦これが行われれば、ある集団と別の集団を分けている突然変異の数を決定することができる。この突然変異の数と、検査された地域のmtDNAの推定 突然変異率から、科学者はMRCA(TMRCA)までのおおよその時間を決定することができる。 Y染色体DNAの場合、TMRCAは異なる方法で算出される。Y-DNAハプログループは、Y-DNAの様々な領域における一塩基多型によって定義され る。ハプログループ内のMRCAに至る時間は、そのハプログループのY染色体のSTR配列にのみ変異が蓄積することによって定義される。非星型クラスター を示すY-STRハプロタイプのY-DNAネットワーク解析は、複数の創始個体によるY-STRの変動を示している。星型クラスターを示す分析は、単一の 祖先から派生した集団を表すとみなすことができる。この場合、Y-STR配列の変動性(マイクロサテライトの変動性とも呼ばれる)は、祖先がこの特定の集 団を創設してからの経過時間の尺度とみなすことができる。チンギス・ハーンまたはその祖先の1人の子孫は、チンギス・ハーンの時代までさかのぼることがで きる有名な星団を代表する[12]。 TMRCAの計算は、世界中に広がった様々な集団の移動年代を決定しようとする際に重要な証拠と見なされる。例えば、ある突然変異が30,000年前に起 こったとみなされた場合、この突然変異はこの日以降に分岐したすべての集団の中で見つかるはずである。考古学的証拠から、文化的伝播と地域的に隔離された 集団の形成が確認された場合、このことは、その後のこの地域における遺伝的突然変異の隔離に反映されなければならない。もし遺伝的分岐と地域的分岐が一致 すれば、考古学的記録によって証明されたように、観察された分岐は移動によるものだと結論づけることができる。しかし、遺伝子の分岐が考古学的記録と異な る時期に起こったのであれば、科学者は遺伝子の分岐を説明するために別の考古学的証拠に目を向けなければならなくなる。この問題は、ヨーロッパの新石器時 代における人口拡散と文化拡散をめぐる論争に最もよく表れている[13]。 現生人類のTMRCA 現生人類のMRCA年代は不明である。母系的MRCAや父系的MRCAの年代よりも必然的に若く、どちらも推定年代はおよそ10万年から20万年前である[14]。 数学者のジョセフ・チャン(Joseph T. Chang)、ダグラス・ローデ(Douglas Rohde)、スティーブ・オルソン(Steve Olson)による研究では、理論モデルを用いて、MRCAが生きていたのは極めて最近、おそらく2,000年ほど前かもしれないと計算している。その結 果、全人類のMRCAはおそらく東アジアに住んでおり、オーストラリアやアメリカ大陸の極めて隔離された個体群に接近する重要な手がかりを与えていただろ うと結論づけた。MRCAが存在した場所としては、アラスカに近いチャクチ半島やカムチャッカ半島、オーストラリアに近いインドネシアやマレーシア、ある いはオーストラリアとアメリカ大陸の中間的な台湾や日本などが考えられる。チャンは、アメリカ大陸とオーストラリアへのヨーロッパ人の植民地化は、 MRCAの年代に実質的な影響を与えるには、あまりにも最近のことであるとしている。実際、もしアメリカ大陸とオーストラリアがヨーロッパ人によって発見 されなかったとしたら、MRCAは現在よりも2.3%ほど過去に遡るだけである[15][16][17]。 ある集団のMRCAの年代は、「最初のカップル」はおろか、集団のボトルネックにも対応していないことに注意してほしい。それはむしろ、過去に高い繁殖成 功を収めた単一の個体が存在し、その遺伝的寄与が時間の経過とともに集団全体に浸透してきたことを反映している。また、MRCAが生きているすべての人に 遺伝情報を伝えたと考えるのも正しくない。有性生殖を通じて、祖先は自分の遺伝子の半分を次の世代の各子孫に受け継ぐ。血統の崩壊がなければ、たった32 世代後には、一人の祖先の寄与は2-32のオーダーになり、ヒトゲノム内の1塩基対以下の数に比例することになる[18]。 |
| Identical ancestors point Main article: Identical ancestors point The MRCA is the most recent common ancestor shared by all individuals in the population under consideration. This MRCA may well have contemporaries who are also ancestral to some but not all of the extant population. The identical ancestors point is a point in the past more remote than the MRCA at which time there are no longer organisms which are ancestral to some but not all of the modern population. Due to pedigree collapse, modern individuals may still exhibit clustering, due to vastly different contributions from each of ancestral population.[19] |
同一祖先ポイント 主な記事 同一祖先点 MRCAとは、対象となる集団の全個体が共有する最も新しい共通祖先のことである。このMRCAには、現存する集団のすべてではないが、ある個体にとって は祖先となる同時代の個体が存在する可能性がある。同一祖先ポイントとは、MRCAよりも遠い過去のある時点のことで、その時点では、現代の集団のすべて ではないが、ある集団の祖先となる生物はもはや存在しない。血統の崩壊により、現代の個体は、それぞれの祖先集団からの寄与が大きく異なるために、依然と してクラスター性を示すことがある[19]。 |
| Cladistics Common descent Coalescent theory, a retrospective model of population genetics Crown group Genealogy, the study of families and the tracing of their lineages and history Genetic distance, the genetic divergence between species or between populations within a species Lowest common ancestor, an analogous concept in graph theory and computer science Phylogenetic tree, a branching diagram or "tree" showing the inferred evolutionary relationships among various biological species Timeline of evolution, outlines the major events in the development of life on the planet Earth Timeline of human evolution, outlines the major events in the development of the human species Last universal common ancestor, the most recent common ancestor of all life |
分類学 共通の子孫 コアレセント理論、集団遺伝学の遡及モデル クラウングループ 家系学:家系を研究し、その系統と歴史をたどること。 遺伝的距離(Genetic distance):種間または種内の集団間の遺伝的乖離。 最小公倍数祖先(Lowest common ancestor):グラフ理論やコンピュータサイエンスにおける類似の概念。 系統樹:様々な生物種間の進化的関係を示す分岐図または「木」。 進化の年表。地球上の生命の発展における主な出来事の概要を示す。 人類進化の年表。ヒトという種の発展における主な出来事の概要を示す。 最後の普遍的共通祖先(Last universal common ancestor):すべての生命の最も最近の共通祖先である。 |
| https://en.wikipedia.org/wiki/Most_recent_common_ancestor |
■ハプロタイプ(haplotype)とは、遺伝子型(ジェノタイプ)の1本(単倍体)の染色体に注目した塩基配列の型。
■類似したハプロタイプを持つ集団のことを「ハプログループ」と呼び、ヒト祖先集団の誕生や移動を推定する研究に活用される。
リ ンク
文 献
そ の他の情報
Copyleft,
CC, Mitzub'ixi Quq Chi'j, 1996-2099