
サンガー・シーケンシング
(第一世代シーケンシング)
Sanger sequencing
☆サ ンガーシークエンシングは、電気泳動法を用いるDNAシークエンシングの手法であり、試験管内でのDNA複製時にDNAポリメラーゼが鎖終結ジデオキシヌ クレオチドをランダムに取り込むことを基盤としている。1977年にフレデリック・サンガーとその同僚によって初めて開発されて以来、約40年にわたって 最も広く使用されているシークエンシング手法となった。スラブゲル電気泳動と蛍光標識を使用する自動化装置は、1987年3月にApplied Biosystems社によって初めて商品化された。[1]その後、自動化スラブゲルは自動キャピラリーアレイ電気泳動に置き換えられた。[2]さらに最 近では、特に大規模な自動ゲノム解析においては、より大容量のサンガーシークエンシングが次世代シークエンシング法に置き換えられている。しかし、サン ガー法は小規模なプロジェクトやディープシーケンスの結果の検証には依然として広く使用されている。 サンガー法は、500ヌクレオチド以上のDNAシーケンスリードを生成でき、99.99%前後の精度で非常に低いエラー率を維持できるという点で、イルミ ナのようなショートリードシーケンス技術よりも優れている。[3] サンガーシーケンスは Sanger法は、SARS-CoV-2のスパイクタンパク質の配列決定[4] などの公衆衛生イニシアティブの取り組みや、疾病対策予防センター(CDC)のCaliciNet監視ネットワークを通じたノロウイルス発生の監視[5] など、現在も積極的に利用されている。
★【関連項目】「サンガー・シーケンシング(第一世代シーケンシング)」→「大規模並列シーケンシング(第二世代シーケンシング)」→「第三世代シーケンシング」→「次世代シーケンシング」
★なお、サンガーシークエンシングの解説の後、関連事項として「マクサム・ギルバート法によるシークエンシング(Maxam–Gilbert sequencing)」を併せて解説する。
| Sanger
sequencing is a method of DNA sequencing that involves electrophoresis
and is based on the random incorporation of chain-terminating
dideoxynucleotides by DNA polymerase during in vitro DNA replication.
After first being developed by Frederick Sanger and colleagues in 1977,
it became the most widely used sequencing method for approximately 40
years. An automated instrument using slab gel electrophoresis and
fluorescent labels was first commercialized by Applied Biosystems in
March 1987.[1] Later, automated slab gels were replaced with automated
capillary array electrophoresis.[2] More recently, higher volume Sanger
sequencing has been replaced by next generation sequencing methods,
especially for large-scale, automated genome analyses. However, the
Sanger method remains in wide use for smaller-scale projects and for
validation of deep sequencing results. It still has the advantage over
short-read sequencing technologies (like Illumina) in that it can
produce DNA sequence reads of > 500 nucleotides and maintains a very
low error rate with accuracies around 99.99%.[3] Sanger sequencing is
still actively being used in efforts for public health initiatives such
as sequencing the spike protein from SARS-CoV-2[4] as well as for the
surveillance of norovirus outbreaks through the Center for Disease
Control and Prevention's (CDC) CaliciNet surveillance network.[5] |
サ
ンガーシークエンシングは、電気泳動法を用いるDNAシークエンシングの手法であり、試験管内でのDNA複製時にDNAポリメラーゼが鎖終結ジデオキシヌ
クレオチドをランダムに取り込むことを基盤としている。1977年にフレデリック・サンガーとその同僚によって初めて開発されて以来、約40年にわたって
最も広く使用されているシークエンシング手法となった。スラブゲル電気泳動と蛍光標識を使用する自動化装置は、1987年3月にApplied
Biosystems社によって初めて商品化された。[1]その後、自動化スラブゲルは自動キャピラリーアレイ電気泳動に置き換えられた。[2]さらに最
近では、特に大規模な自動ゲノム解析においては、より大容量のサンガーシークエンシングが次世代シークエンシング法に置き換えられている。しかし、サン
ガー法は小規模なプロジェクトやディープシーケンスの結果の検証には依然として広く使用されている。
サンガー法は、500ヌクレオチド以上のDNAシーケンスリードを生成でき、99.99%前後の精度で非常に低いエラー率を維持できるという点で、イルミ
ナのようなショートリードシーケンス技術よりも優れている。[3] サンガーシーケンスは
Sanger法は、SARS-CoV-2のスパイクタンパク質の配列決定[4]
などの公衆衛生イニシアティブの取り組みや、疾病対策予防センター(CDC)のCaliciNet監視ネットワークを通じたノロウイルス発生の監視[5]
など、現在も積極的に利用されている。 |
Method Fluorescent ddNTP molecules The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotide triphosphates (dNTPs), and modified di-deoxynucleotide triphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines. The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones. The deoxynucleotide concentration should be approximately 100-fold higher than that of the corresponding dideoxynucleotide (e.g. 0.5mM dTTP : 0.005mM ddTTP) to allow enough fragments to be produced while still transcribing the complete sequence (but the concentration of ddNTP also depends on the desired length of sequence).[6] Putting it in a more sensible order, four separate reactions are needed in this process to test all four ddNTPs. Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. In the original publication of 1977,[6] the formation of base-paired loops of ssDNA was a cause of serious difficulty in resolving bands at some locations. This is frequently performed using a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C). The DNA bands may then be visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image. 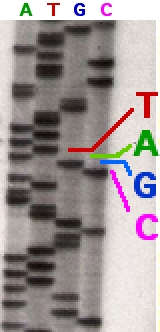 Part of a radioactively labelled sequencing gel In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes, from bottom to top, are then used to read the DNA sequence. 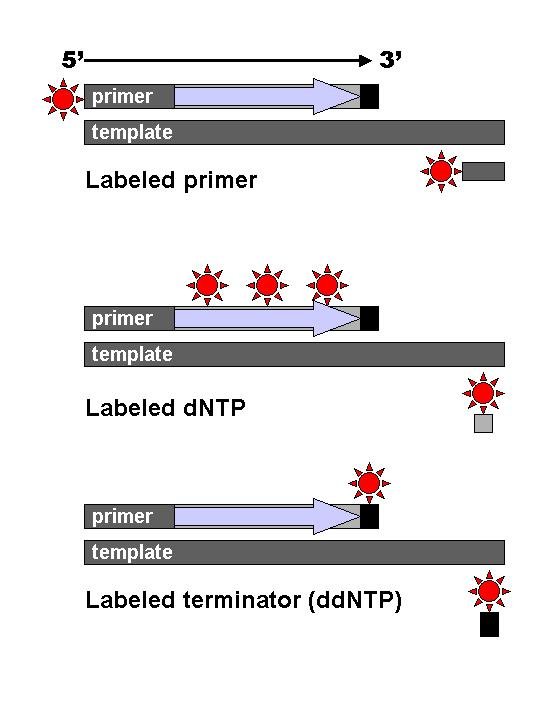 DNA fragments are labelled with a radioactive or fluorescent tag on the primer (1), in the new DNA strand with a labeled dNTP, or with a labeled ddNTP. Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5' end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers[7][8] of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing. 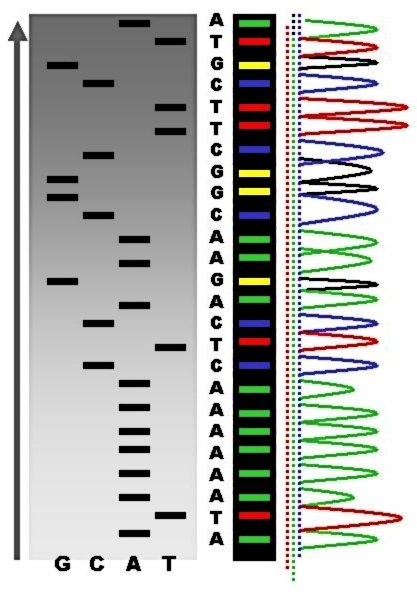 Sequence ladder by radioactive sequencing compared to fluorescent peaks Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence. Dye-terminator sequencing 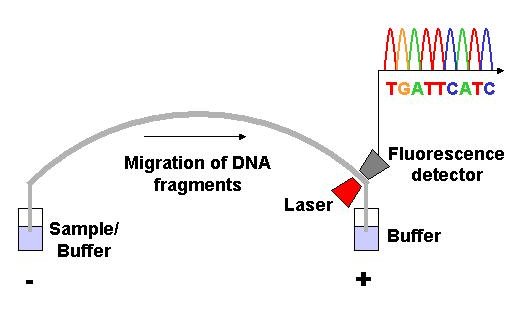 Capillary electrophoresis Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which emits light at different wavelengths. Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing. Its limitations include dye effects due to differences in the incorporation of the dye-labelled chain terminators into the DNA fragment, resulting in unequal peak heights and shapes in the electronic DNA sequence trace electropherogram (a type of chromatogram) after capillary electrophoresis (see figure to the left). This problem has been addressed with the use of modified DNA polymerase enzyme systems and dyes that minimize incorporation variability, as well as methods for eliminating "dye blobs". The dye-terminator sequencing method, along with automated high-throughput DNA sequence analyzers, was used for the vast majority of sequencing projects until the introduction of next generation sequencing. Automation and sample preparation  View of the start of an example dye-terminator read Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch. Batch runs may occur up to 24 times a day. DNA sequencers separate strands by size (or length) using capillary electrophoresis, they detect and record dye fluorescence, and output data as fluorescent peak trace chromatograms. Sequencing reactions (thermocycling and labelling), cleanup and re-suspension of samples in a buffer solution are performed separately, before loading samples onto the sequencer. A number of commercial and non-commercial software packages can trim low-quality DNA traces automatically. These programs score the quality of each peak and remove low-quality base peaks (which are generally located at the ends of the sequence).[9] The accuracy of such algorithms is inferior to visual examination by a human operator, but is adequate for automated processing of large sequence data sets. Applications of dye-terminating sequencing The field of public health plays many roles to support patient diagnostics as well as environmental surveillance of potential toxic substances and circulating biological pathogens. Public health laboratories (PHL) and other laboratories around the world have played a pivotal role in providing rapid sequencing data for the surveillance of the virus SARS-CoV-2, causative agent for COVID-19, during the pandemic that was declared a public health emergency on January 30, 2020.[10] Laboratories were tasked with the rapid implementation of sequencing methods and asked to provide accurate data to assist in the decision-making models for the development of policies to mitigate spread of the virus. Many laboratories resorted to next generation sequencing methodologies while others supported efforts with Sanger sequencing. The sequencing efforts of SARS-CoV-2 are many, while most laboratories implemented whole genome sequencing of the virus, others have opted to sequence very specific genes of the virus such as the S-gene, encoding the information needed to produce the spike protein. The high mutation rate of SARS-CoV-2 leads to genetic differences within the S-gene and these differences have played a role in the infectivity of the virus.[11] Sanger sequencing of the S-gene provides a quick, accurate, and more affordable method to retrieving the genetic code. Laboratories in lower income countries may not have the capabilities to implement expensive applications such as next generation sequencing, so Sanger methods may prevail in supporting the generation of sequencing data for surveillance of variants. Sanger sequencing is also the "gold standard" for norovirus surveillance methods for the Center for Disease Control and Prevention's (CDC) CaliciNet network. CalciNet is an outbreak surveillance network that was established in March 2009. The goal of the network is to collect sequencing data of circulating noroviruses in the United States and activate downstream action to determine the source of infection to mitigate the spread of the virus. The CalciNet network has identified many infections as foodborne illnesses.[5] This data can then be published and used to develop recommendations for future action to prevent tainting food. The methods employed for detection of norovirus involve targeted amplification of specific areas of the genome. The amplicons are then sequenced using dye-terminating Sanger sequencing and the chromatograms and sequences generated are analyzed with a software package developed in BioNumerics. Sequences are tracked and strain relatedness is studied to infer epidemiological relevance. Challenges Common challenges of DNA sequencing with the Sanger method include poor quality in the first 15-40 bases of the sequence due to primer binding and deteriorating quality of sequencing traces after 700-900 bases. Base calling software such as Phred typically provides an estimate of quality to aid in trimming of low-quality regions of sequences.[12][13] In cases where DNA fragments are cloned before sequencing, the resulting sequence may contain parts of the cloning vector. In contrast, PCR-based cloning and next-generation sequencing technologies based on pyrosequencing often avoid using cloning vectors. Recently, one-step Sanger sequencing (combined amplification and sequencing) methods such as Ampliseq and SeqSharp have been developed that allow rapid sequencing of target genes without cloning or prior amplification.[14][15] Current methods can directly sequence only relatively short (300-1000 nucleotides long) DNA fragments in a single reaction. The main obstacle to sequencing DNA fragments above this size limit is insufficient power of separation for resolving large DNA fragments that differ in length by only one nucleotide. |
方法 蛍光性ddNTP分子 古典的な鎖終結法では、一本鎖DNAテンプレート、DNAプライマー、DNAポリメラーゼ、通常のデオキシヌクレオチド三リン酸(dNTP)、および修飾 ジデオキシヌクレオチド三リン酸(ddNTP)が必要である。後者はDNA鎖伸長を終結させる。これらの鎖終結ヌクレオチドは、2つのヌクレオチド間のリ ン酸ジエステル結合の形成に必要な3'-OH基を持たないため、修飾ddNTPが組み込まれるとDNAポリメラーゼによるDNAの伸長が停止する。 ddNTPは、自動シークエンシング装置での検出用に、放射性または蛍光標識される場合がある。 DNAサンプルは4つの別々の配列反応に分けられ、4つの標準デオキシヌクレオチド(dATP、dGTP、dCTP、dTTP)とDNAポリメラーゼがす べて含まれる。各反応には4つのジデオキシヌクレオチド(ddATP、ddGTP、ddCTP、またはddTTP)のうちの1つだけが加えられ、他の添加 ヌクレオチドは通常のヌクレオチドである。デオキシヌクレオチドの濃度は、対応するジデオキシヌクレオチドの濃度の約100倍(例えば、0.5mMの dTTP:0.005mMのddTTP)であるべきである。完全な配列を転写しながら、十分な断片が生成されるようにするためである(ただし、ddNTP の濃度は、目的とする配列の長さにも依存する)。[6] よりわかりやすい順序で説明すると、このプロセスでは4つのddNTPすべてをテストするために4つの別々の反応が必要である。結合したプライマーからの テンプレートDNAの伸長反応が複数回行われた後、生成されたDNA断片は熱変性され、ゲル電気泳動によってサイズ別に分離される。1977年の最初の論 文では、ssDNAの塩基対ループの形成が、いくつかの位置でバンドを分離する際に深刻な問題を引き起こしていた。これは、変性ポリアクリルアミド尿素ゲ ルを使用して、4つの反応を4つの個別のレーン(レーンA、T、G、C)で実行することで、頻繁に行われる。その後、DNAバンドはオートラジオグラ フィーまたはUV光で可視化され、DNA配列はX線フィルムまたはゲル画像から直接読み取ることができる。 放射性標識シーケンシングゲルの一部 右の画像では、X線フィルムがゲルに露光されており、暗いバンドは異なる長さのDNA断片に対応している。レーン内の暗いバンドは、ジデオキシヌクレオチ ド(ddATP、ddGTP、ddCTP、またはddTTP)の取り込み後の鎖終結の結果生じたDNA断片を示す。 4つのレーンにおける異なるバンドの相対的な位置(下から上)が、DNA配列の解読に使用される。 DNA断片は、プライマー(1)に放射性または蛍光標識を付けたり、標識dNTPを付けた新しいDNA鎖、または標識ddNTPを付けたものとなる。 鎖停止シーケンシングの技術的バリエーションには、放射性リンを含むヌクレオチドによる標識付け(放射標識)や、5'末端に蛍光色素で標識したプライマー を使用する方法などがある。 蛍光色素プライマーシーケンシングは、光学システムでの読み取りを容易にし、より迅速で経済的な分析と自動化を実現する。 Leroy Hoodと共同研究者らによる蛍光標識ddNTPとプライマーの開発[7][8]は、自動化された高処理DNAシーケンシングの基礎を築いた。 放射性シークエンシングによるシークエンスラダーと蛍光ピークの比較 鎖停止法は、DNAシークエンシングを大幅に簡素化した。例えば、シークエンシングに必要な試薬がすべて含まれ、あらかじめ分注され、すぐに使える鎖停止 法に基づくキットが市販されている。 制限事項としては、プライマーのDNAへの非特異的結合がDNAシークエンスの正確な読み取りに影響を与えること、およびDNAの二次構造がシークエンス の正確性に影響を与えることが挙げられる。 色素終結法シークエンシング キャピラリー電気泳動 ダイターミネーターシークエンシングでは、鎖終結体であるddNTPの標識を利用する。これにより、標識プライマー法のように4回の反応ではなく、1回の 反応でシークエンシングが可能となる。ダイターミネーターシークエンシングでは、4種類のジデオキシヌクレオチド鎖終結体のそれぞれに蛍光色素で標識を付 ける。 その利便性とスピードの良さから、現在では、ダイターミネーターシークエンシングが自動シークエンシングの主流となっている。 その限界としては、DNA断片へのダイ標識鎖ターミネーターの組み込みの差異による色素効果があり、キャピラリー電気泳動後の電子DNA配列トレース電気 泳動図(クロマトグラムの一種)において、ピークの高さや形状が不均一になる(左図参照)。 この問題は、取り込みのばらつきを最小限に抑えるよう改良されたDNAポリメラーゼ酵素システムや色素、および「色素塊」を除去する方法によって対処され ている。色素終結子シーケンス法は、自動化された高処理DNAシーケンス分析装置とともに、次世代シーケンスが導入されるまで、シーケンスプロジェクトの 大半で使用されていた。 自動化とサンプル準備 色素終結反応の読み取りの開始の様子 自動DNAシークエンサー(DNAシークエンサー)は、1回のバッチで最大384のDNAサンプルをシークエンシングできる。バッチ処理は1日最大24回 実施される。DNAシークエンサーは、キャピラリー電気泳動法を用いて鎖をサイズ(または長さ)によって分離し、色素の蛍光を検出し記録し、蛍光ピークト レースクロマトグラムとしてデータを出力する。シークエンサーにサンプルをロードする前に、シークエンシング反応(サーマルサイクリングおよび標識)、サ ンプルの洗浄、緩衝液中への再懸濁は個別に実行される。商業用および非商業用のソフトウェアパッケージの多くは、低品質のDNAトレースを自動的にトリミ ングすることができる。これらのプログラムは各ピークの品質をスコア化し、低品質の塩基ピーク(通常は配列の末端に位置する)を除去する。[9] このようなアルゴリズムの精度は、人間のオペレーターによる目視検査には劣るが、大量の配列データの自動処理には十分である。 色素終結シーケンシングの応用 公衆衛生の分野では、患者の診断だけでなく、潜在的な有毒物質や循環する生物学的病原体の環境監視もサポートする多くの役割を担っている。公衆衛生研究所 (PHL)や世界中のその他の研究所は、2020年1月30日に公衆衛生上の緊急事態が宣言されたパンデミックのさなか、新型コロナウイルス感染症 (COVID-19)の原因となるウイルスSARS-CoV-2の監視に迅速なシーケンスデータを提供するという重要な役割を果たした 。 2020年1月30日に公衆衛生上の緊急事態が宣言されたパンデミックのさなか、ラボは迅速なシーケンス手法の実施を求められ、ウイルスの蔓延を抑制する ための政策策定の意思決定モデルを支援する正確なデータの提供を求められた。多くの研究所は次世代シーケンシング法を採用したが、サンガー法によるシーケ ンシングで対応する研究所もあった。 SARS-CoV-2のシーケンシングには多くの手法が用いられているが、ほとんどの研究所はウイルスの全ゲノムシーケンシングを実施している。一方で、 スパイクタンパク質の生成に必要な情報をコード化するS遺伝子など、ウイルスの特定の遺伝子のみをシーケンシングする研究所もある。SARS-CoV-2 の変異率が高いことから、S-遺伝子内に遺伝子差異が生じ、この差異がウイルスの感染力に影響を与えている。[11] S-遺伝子のサンガーシークエンシングは、遺伝暗号の取得に迅速かつ正確で、より手頃な方法である。低所得国の研究所では、次世代シークエンシングのよう な高価なアプリケーションを導入する能力がない場合があるため、変異株の監視のためのシークエンシングデータの生成をサポートする上で、サンガー法が主流 となる可能性がある。 また、サンガーシークエンシングは、米国疾病対策センター(CDC)のCaliciNetネットワークにおけるノロウイルス監視方法の「ゴールドスタン ダード」でもある。CalciNetは2009年3月に設立された感染症発生動向調査ネットワークである。このネットワークの目的は、米国で流行している ノロウイルスのシークエンシングデータを収集し、感染源を特定してウイルスの蔓延を抑制するための下流の行動を活性化することである。CalciNet ネットワークは、多くの感染が食品由来であることを特定した。[5] このデータは公表され、食品汚染を防ぐための今後の対策の推奨事項を策定するために使用される。 ノロウイルスの検出に使用される手法は、ゲノムの特定領域の標的増幅を含む。 増幅産物は、色素終結サンガーシークエンシングを使用してシークエンシングされ、生成されたクロマトグラムと配列は、BioNumericsで開発された ソフトウェアパッケージで分析される。配列は追跡され、株の関連性が研究され、疫学的な関連性が推測される。 課題 サンガー法によるDNAシークエンシングの一般的な課題には、プライマーの結合によるシークエンスの最初の15~40塩基の質の悪さ、および 700~900塩基以降のシークエンストレースの質の悪化が含まれる。Phredなどのベースコーリングソフトウェアは通常、シークエンスの低品質領域の トリミングを支援するために、品質の推定値を提供する。[12][13] シーケンス前にDNA断片がクローニングされる場合、結果として得られる配列にはクローニングベクターの部分が含まれる可能性がある。これに対し、PCR ベースのクローニングやパイロシークエンシングに基づく次世代シーケンシング技術では、クローニングベクターを使用しないことが多い。最近では、 AmpliseqやSeqSharpなどのワンステップサンガーシーケンス(増幅とシーケンスを組み合わせた)法が開発されており、クローニングや事前の 増幅を行わずに標的遺伝子のシーケンスを迅速に行うことができる。 現在の方法では、1回の反応で直接配列決定できるのは比較的短い(長さ300~1000ヌクレオチド)DNA断片のみである。このサイズ制限を超える DNA断片の配列決定における主な障害は、長さが1ヌクレオチドしか違わない大きなDNA断片を分離する際に、分離能力が不十分であることである。 |
| Microfluidic Sanger sequencing Microfluidic Sanger sequencing is a lab-on-a-chip application for DNA sequencing, in which the Sanger sequencing steps (thermal cycling, sample purification, and capillary electrophoresis) are integrated on a wafer-scale chip using nanoliter-scale sample volumes. This technology generates long and accurate sequence reads, while obviating many of the significant shortcomings of the conventional Sanger method (e.g. high consumption of expensive reagents, reliance on expensive equipment, personnel-intensive manipulations, etc.) by integrating and automating the Sanger sequencing steps. In its modern inception, high-throughput genome sequencing involves fragmenting the genome into small single-stranded pieces, followed by amplification of the fragments by polymerase chain reaction (PCR). Adopting the Sanger method, each DNA fragment is irreversibly terminated with the incorporation of a fluorescently labeled dideoxy chain-terminating nucleotide, thereby producing a DNA “ladder” of fragments that each differ in length by one base and bear a base-specific fluorescent label at the terminal base. Amplified base ladders are then separated by capillary array electrophoresis (CAE) with automated, in situ “finish-line” detection of the fluorescently labeled ssDNA fragments, which provides an ordered sequence of the fragments. These sequence reads are then computer assembled into overlapping or contiguous sequences (termed "contigs") which resemble the full genomic sequence once fully assembled.[16] Sanger methods achieve maximum read lengths of approximately 800 bp (typically 500–600 bp with non-enriched DNA). The longer read lengths in Sanger methods display significant advantages over other sequencing methods especially in terms of sequencing repetitive regions of the genome. A challenge of short-read sequence data is particularly an issue in sequencing new genomes (de novo) and in sequencing highly rearranged genome segments, typically those seen of cancer genomes or in regions of chromosomes that exhibit structural variation.[17] Applications of microfluidic sequencing technologies Other useful applications of DNA sequencing include single nucleotide polymorphism (SNP) detection, single-strand conformation polymorphism (SSCP) heteroduplex analysis, and short tandem repeat (STR) analysis. Resolving DNA fragments according to differences in size and/or conformation is the most critical step in studying these features of the genome.[16] Device design The sequencing chip has a four-layer construction, consisting of three 100-mm-diameter glass wafers (on which device elements are microfabricated) and a polydimethylsiloxane (PDMS) membrane. Reaction chambers and capillary electrophoresis channels are etched between the top two glass wafers, which are thermally bonded. Three-dimensional channel interconnections and microvalves are formed by the PDMS and bottom manifold glass wafer. The device consists of three functional units, each corresponding to the Sanger sequencing steps. The thermal cycling (TC) unit is a 250-nanoliter reaction chamber with integrated resistive temperature detector, microvalves, and a surface heater. Movement of reagent between the top all-glass layer and the lower glass-PDMS layer occurs through 500-μm-diameter via-holes. After thermal-cycling, the reaction mixture undergoes purification in the capture/purification chamber, and then is injected into the capillary electrophoresis (CE) chamber. The CE unit consists of a 30-cm capillary which is folded into a compact switchback pattern via 65-μm-wide turns. Sequencing chemistry Thermal cycling In the TC reaction chamber, dye-terminator sequencing reagent, template DNA, and primers are loaded into the TC chamber and thermal-cycled for 35 cycles ( at 95 °C for 12 seconds and at 60 °C for 55 seconds). Purification The charged reaction mixture (containing extension fragments, template DNA, and excess sequencing reagent) is conducted through a capture/purification chamber at 30 °C via a 33-Volts/cm electric field applied between capture outlet and inlet ports. The capture gel through which the sample is driven, consists of 40 μM of oligonucleotide (complementary to the primers) covalently bound to a polyacrylamide matrix. Extension fragments are immobilized by the gel matrix, and excess primer, template, free nucleotides, and salts are eluted through the capture waste port. The capture gel is heated to 67-75 °C to release extension fragments. Capillary electrophoresis Extension fragments are injected into the CE chamber where they are electrophoresed through a 125-167-V/cm field. Platforms The Apollo 100 platform (Microchip Biotechnologies Inc., Dublin, CA)[18] integrates the first two Sanger sequencing steps (thermal cycling and purification) in a fully automated system. The manufacturer claims that samples are ready for capillary electrophoresis within three hours of the sample and reagents being loaded into the system. The Apollo 100 platform requires sub-microliter volumes of reagents. Comparisons to other sequencing techniques Performance values for genome sequencing technologies including Sanger methods and next-generation methods[17][19][20] 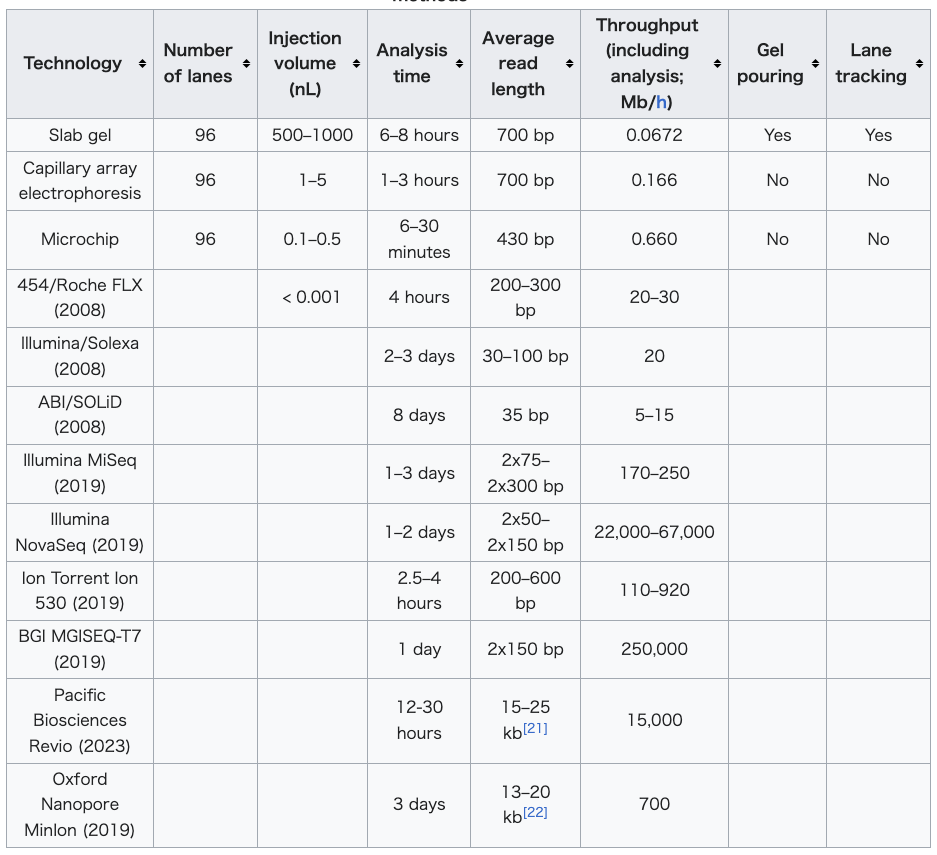 |
マイクロフルイディクス・サンガーシークエンシング マイクロフルイディクス・サンガーシークエンシングは、DNAシークエンシングのためのラボオンチップアプリケーションであり、サンガーシークエンシング のステップ(サーマルサイクリング、サンプル精製、キャピラリー電気泳動)をナノリットル単位のサンプル量を使用してウェハースケールのチップに統合す る。この技術は、長い正確な配列の読み取りを生成する一方で、サンガーシークエンシングのステップを統合し自動化することで、従来のサンガー法の重大な欠 点(高価な試薬の大量消費、高価な機器への依存、人手を要する操作など)の多くを解消している。 現代的な手法では、ゲノムのハイスループットシークエンシングでは、ゲノムを小さな一本鎖断片に断片化し、続いてポリメラーゼ連鎖反応(PCR)により断 片を増幅する。サンガー法では、各DNA断片は蛍光標識されたジデオキシ鎖終結ヌクレオチドが組み込まれることで不可逆的に終結し、その結果、長さが1塩 基ずつ異なる断片のDNA「ラダー」が生成される。増幅された塩基ラダーは、キャピラリーアレイ電気泳動(CAE)によって分離され、蛍光標識された一本 鎖DNA断片が自動的にその場で検出され、断片の順序配列が得られる。これらの配列は、コンピュータによって重複または連続する配列(「コンティグ」と呼 ばれる)に組み立てられ、完全に組み立てられたゲノム配列全体に似たものとなる。 サンガー法では、最大リード長は約800bp(通常は非濃縮DNAで500~600bp)である。サンガー法による長いリード長は、特にゲノムの反復領域 のシーケンスという点において、他のシーケンス法よりも大きな利点がある。短いリード長のシーケンスデータの課題は、特に新規ゲノムのシーケンス(デノ ボ)や、高度に再編成されたゲノムセグメントのシーケンス、通常は癌ゲノムや構造変化を示す染色体領域のシーケンスにおいて問題となる。 マイクロ流体シーケンシング技術の応用 DNAシーケンシングのその他の有用な応用例としては、一塩基多型(SNP)の検出、一本鎖構造多型(SSCP)ヘテロ二重鎖分析、および短鎖反復配列 (STR)分析などがある。 サイズおよび/または構造の違いに基づいてDNA断片を解明することは、ゲノムのこれらの特徴を研究する上で最も重要なステップである。 装置設計 シーケンシングチップは4層構造で、直径100mmのガラスウエハ3枚(デバイス要素が微細加工されている)とポリジメチルシロキサン(PDMS)膜で構 成されている。反応室とキャピラリー電気泳動チャンネルは、熱結合された上部の2枚のガラスウエハの間にエッチングされている。3次元チャンネルの相互接 続とマイクロバルブは、PDMSと下部のマニフォールドガラスウエハによって形成されている。 この装置は、サンガーシークエンシングの各ステップに対応する3つの機能ユニットで構成されている。サーマルサイクリング(TC)ユニットは、抵抗温度検 出器、マイクロバルブ、表面ヒーターが統合された250ナノリットルの反応チャンバーである。上部オールガラス層と下部ガラス-PDMS層間の試薬の移動 は、直径500μmのバイアホールを介して行われる。熱サイクル後、反応混合物はキャプチャー/精製チャンバーで精製され、その後キャピラリー電気泳動 (CE)チャンバーに注入される。CEユニットは、65μm幅のターンでコンパクトなスイッチバックパターンに折りたたまれた30cmのキャピラリーで構 成されている。 シーケンス化学 熱サイクル TC反応チャンバーに、色素ターミネーターシーケンス試薬、テンプレートDNA、プライマーをロードし、35サイクルの熱サイクルを行う(95℃で12秒間、60℃で55秒間)。 精製 電荷を帯びた反応混合液(伸長断片、テンプレートDNA、過剰のシーケンス試薬を含む)は、キャプチャー用アウトレットとインレットポートの間に印加され た33ボルト/cmの電界により、30℃でキャプチャー/精製チャンバーを通過する。サンプルが通過するキャプチャーゲルは、40μMのオリゴヌクレオチ ド(プライマーと相補的)がポリアクリルアミドマトリックスに共有結合したものから構成される。伸長断片はゲルマトリックスに固定化され、余剰のプライ マー、鋳型、遊離ヌクレオチド、塩類はキャプチャー廃液ポートから溶出される。 キャプチャーゲルは67~75℃に加熱され、伸長断片が放出される。 キャピラリー電気泳動 伸長断片はCEチャンバーに注入され、125~167 V/cmの電場により電気泳動される。 プラットフォーム Apollo 100 プラットフォーム(Microchip Biotechnologies Inc.、カリフォルニア州ダブリン)[18] は、最初の2つのサンガーシークエンシング工程(サーマルサイクリングと精製)を完全に自動化されたシステムに統合している。メーカーの主張によると、サ ンプルと試薬をシステムにロードしてから3時間以内に、サンプルをキャピラリー電気泳動に供することができる。Apollo 100 プラットフォームでは、サブマイクロリットルの試薬量が必要である。 他のシーケンシング技術との比較 サンガー法や次世代シーケンサー法を含むゲノムシーケンシング技術の性能値[17][19][20] |
| The
ultimate goal of high-throughput sequencing is to develop systems that
are low-cost, and extremely efficient at obtaining extended (longer)
read lengths. Longer read lengths of each single electrophoretic
separation, substantially reduces the cost associated with de novo DNA
sequencing and the number of templates needed to sequence DNA contigs
at a given redundancy. Microfluidics may allow for faster, cheaper and
easier sequence assembly.[16] |
ハ
イスループットシーケンシングの究極の目標は、低コストで、より長いリード長を極めて効率的に取得できるシステムを開発することである。電気泳動分離の
リード長が長くなるほど、de novo
DNAシーケンシングにかかるコストと、所定の冗長性でDNAコンティグをシーケンシングする際に必要なテンプレートの数が大幅に削減される。マイクロフ
ルイディクスは、より高速で安価かつ容易なシーケンスアセンブリを可能にするかもしれない。[16] |
| Maxam–Gilbert sequencing Second-generation sequencing Third-generation sequencing |
マクサム・ギルバート法によるシークエンシング 第二世代シークエンシング(大規模並列シーケンシング) 第三世代シークエンシング |
Maxam–Gilbert sequencing
is a method of DNA sequencing developed by Allan Maxam and Walter
Gilbert in 1976–1977. This method is based on nucleobase-specific
partial chemical modification of DNA and subsequent cleavage of the DNA
backbone at sites adjacent to the modified nucleotides.[1] An example Maxam–Gilbert sequencing reaction. Cleaving the same tagged segment of DNA at different points yields tagged fragments of different sizes. The fragments may then be separated by gel electrophoresis. Maxam–Gilbert sequencing was the first widely adopted method for DNA sequencing, and, along with the Sanger dideoxy method, represents the first generation of DNA sequencing methods. Maxam–Gilbert sequencing is no longer in widespread use, having been supplanted by next-generation sequencing methods. History Although Maxam and Gilbert published their chemical sequencing method two years after Frederick Sanger and Alan Coulson published their work on plus-minus sequencing,[2][3] Maxam–Gilbert sequencing rapidly became more popular, since purified DNA could be used directly, while the initial Sanger method required that each read start be cloned for production of single-stranded DNA. However, with the improvement of the chain-termination method (see below), Maxam–Gilbert sequencing has fallen out of favour due to its technical complexity prohibiting its use in standard molecular biology kits, extensive use of hazardous chemicals, and difficulties with scale-up.[4] Allan Maxam and Walter Gilbert’s 1977 paper “A new method for sequencing DNA” was honored by a Citation for Chemical Breakthrough Award from the Division of History of Chemistry of the American Chemical Society for 2017. It was presented to the Department of Molecular & Cellular Biology, Harvard University.[5] Procedure Maxam–Gilbert sequencing requires radioactive labeling at one 5′ end of the DNA fragment to be sequenced (typically by a kinase reaction using gamma-32P ATP) and purification of the DNA. Chemical treatment generates breaks at a small proportion of one or two of the four nucleotide bases in each of four reactions (G, A+G, C, C+T). For example, the purines (A+G) are depurinated using formic acid, the guanines (and to some extent the adenines) are methylated by dimethyl sulfate, and the pyrimidines (C+T) are hydrolysed using hydrazine. The addition of salt (sodium chloride) to the hydrazine reaction inhibits the reaction of thymine for the C-only reaction. The modified DNAs may then be cleaved by hot piperidine; (CH2)5NH at the position of the modified base. The concentration of the modifying chemicals is controlled to introduce on average one modification per DNA molecule. Thus a series of labeled fragments is generated, from the radiolabeled end to the first "cut" site in each molecule. The fragments in the four reactions are electrophoresed side by side in denaturing acrylamide gels for size separation. To visualize the fragments, the gel is exposed to X-ray film for autoradiography, yielding a series of dark bands each showing the location of identical radiolabeled DNA molecules. From presence and absence of certain fragments the sequence may be inferred.[1][6] Related methods This method led to the Methylation Interference Assay, used to map DNA-binding sites for DNA-binding proteins.[7] An automated Maxam–Gilbert sequencing protocol was developed in 1994.[8] |
マクサム・ギルバート法は、
1976年から1977年にかけてアラン・マクサムとウォルター・ギルバートによって開発されたDNA塩基配列決定法である。この方法は、DNAの核酸塩
基特異的な部分化学修飾と、その後に続く修飾されたヌクレオチドに隣接する部位でのDNA骨格の切断に基づいている。 Maxam-Gilbert法によるシークエンス反応の例。 同じタグ付きDNA断片を異なる位置で切断すると、異なるサイズのタグ付き断片が得られる。 断片はゲル電気泳動により分離できる。 Maxam-Gilbert法は、DNAシークエンス法として初めて広く採用された方法であり、サンガーのジデオキシ法とともに、DNAシークエンス法の 第一世代を代表するものである。 Maxam-Gilbert法は、次世代シークエンス法に取って代わられ、現在では広く使用されることはなくなった。 歴史 マクサムとギルバートが化学的シークエンシング法を発表したのは、フレデリック・サンガーとアラン・カールソンがプラス・マイナスシークエンシング法を発 表してから2年後のことだったが[2][3]、精製したDNAを直接使用できるマクサム・ギルバート法は急速に普及した。一方、初期のサンガー法では、一 本鎖DNAの生成には各リードの開始部分をクローニングする必要があった。しかし、連鎖停止法(下記参照)の改良により、マクサム・ギルバート法は技術的 に複雑であるため標準的な分子生物学キットでの使用が禁止され、また危険な化学物質を大量に使用することや、スケールアップが難しいことなどから、次第に 用いられなくなった。 アラン・マクサムとウォルター・ギルバートの1977年の論文「DNAの配列決定のための新しい方法」は、2017年のアメリカ化学会の化学史部門から化学的進歩賞を授与された。 ハーバード大学分子細胞生物学部に贈られた。[5] 手順 Maxam-Gilbert法では、配列決定するDNA断片の5'末端を放射性標識する必要がある(通常はガンマ線32P-ATPを用いたキナーゼ反応に よる)。化学処理により、4つの反応(G、A+G、C、C+T)それぞれにおける4つのヌクレオチド塩基のうち、1つまたは2つのごく一部に切断が生じ る。例えば、プリン塩基(A+G)はギ酸で脱プリン化され、グアニン塩基(およびある程度アデニン塩基)はジメチル硫酸でメチル化され、ピリミジン塩基 (C+T)はヒドラジンで加水分解される。C-only反応では、ヒドラジン反応に塩(塩化ナトリウム)を加えることでチミン塩基の反応が阻害される。そ の後、修飾されたDNAは、修飾された塩基の位置でピペリジン(CH2)5NHによって切断される。修飾化学物質の濃度は、DNA分子あたり平均1つの修 飾を導入するように制御される。これにより、放射性標識された末端から各分子の最初の「切断」部位まで、一連の標識された断片が生成される。 4つの反応で生成された断片は、サイズ分離のために変性アクリルアミドゲルで並べて電気泳動される。断片を可視化するために、ゲルをオートラジオグラ フィー用のX線フィルムに露光し、同一の放射性標識DNA分子の位置を示す一連の暗いバンドが得られる。特定の断片の有無から、配列を推測することができ る。[1][6] 関連方法 この方法は、DNA結合タンパク質のDNA結合部位をマッピングするために使用されるメチル化干渉アッセイにつながった。[7] 1994年には、自動化Maxam-Gilbertシークエンシングプロトコルが開発された。[8] |
| https://en.wikipedia.org/wiki/Maxam%E2%80%93Gilbert_sequencing |
|
リ ンク
文 献
そ の他の情報
Copyleft,
CC, Mitzub'ixi Quq Chi'j, 1996-2099