
次世代シーケンシング
Next-Generation Sequencing (NGS), or
Massive parallel sequencing (MGS)
☆ 次世代シーケンシング(NGS)、大規模並列シーケンシング、ディープシーケンシングは、ゲノム研究に革命をもたらしたDNAシーケンシング技術を説明す る関連用語である。NGSを使用すれば、ヒトゲノム全体を1日以内に配列決定することができる。これに対し、ヒトゲノム解読に使用された従来のサンガー シークエンシング技術では、最終草案の完成までに10年以上を要した。ゲノム研究では、NGSが従来のサンガーシークエンシングに取って代わるケースがほ とんどであるが、臨床現場での日常的な使用にはまだ至っていない(→「ゲノミクス」)。
★以下は、まずは、この世代の直前の、第三世代シークエンシングについて解説する。
★【関連項目】「サンガー・シーケンシング(第一世代シーケンシング)」→「大規模並列シーケンシング(第二世代シーケンシング)」→「第三世代シーケンシング」→「次世代シーケンシング」
| Third-generation
sequencing (also known as long-read sequencing) is a class of DNA
sequencing methods which produce longer sequence reads, under active
development since 2008.[1] Third generation sequencing technologies have the capability to produce substantially longer reads than second generation sequencing, also known as next-generation sequencing.[1] Such an advantage has critical implications for both genome science and the study of biology in general. However, third generation sequencing data have much higher error rates than previous technologies, which can complicate downstream genome assembly and analysis of the resulting data.[2] These technologies are undergoing active development and it is expected that there will be improvements to the high error rates. For applications that are more tolerant to error rates, such as structural variant calling, third generation sequencing has been found to outperform existing methods, even at a low depth of sequencing coverage.[3] Current technologies Sequencing technologies with a different approach than second-generation platforms were first described as "third-generation" in 2008–2009.[4] There are several companies currently at the heart of third generation sequencing technology development, namely, Pacific Biosciences, Oxford Nanopore Technology, Quantapore (CA-USA), and Stratos (WA-USA). These companies are taking fundamentally different approaches to sequencing single DNA molecules. PacBio developed the sequencing platform of single molecule real time sequencing (SMRT), based on the properties of zero-mode waveguides. Signals are in the form of fluorescent light emission from each nucleotide incorporated by a DNA polymerase bound to the bottom of the zL well. Oxford Nanopore’s technology involves passing a DNA molecule through a nanoscale pore structure and then measuring changes in electrical field surrounding the pore; while Quantapore has a different proprietary nanopore approach. Stratos Genomics spaces out the DNA bases with polymeric inserts, "Xpandomers", to circumvent the signal to noise challenge of nanopore ssDNA reading. Also notable is Helicos's single molecule fluorescence approach, but the company entered bankruptcy in the fall of 2015. |
第3世代シーケンシング(ロングリードシーケンシングとも呼ばれる)は、より長いシーケンスリードを生成するDNAシーケンシング手法の一種であり、2008年より活発に開発が進められている。 第3世代シーケンシング技術は、第2世代シーケンシングよりもはるかに長いリードを生成する能力があり、次世代シーケンシングとも呼ばれる。このような利 点は、ゲノム科学および生物学全般の研究にとって非常に重要な意味を持つ。しかし、第3世代シーケンシングデータは、以前の技術よりもエラー率がはるかに 高く、その結果、下流のゲノムアセンブリやデータ解析が複雑になる可能性がある。[2] これらの技術は現在も活発に開発が進められており、エラー率の改善が期待されている。 構造多型検出など、エラー率に寛容なアプリケーションでは、シーケンスカバレッジの深度が低い場合でも、第3世代シーケンシングが既存の方法よりも優れた 性能を発揮することが分かっている。[3] 現在の技術 第二世代プラットフォームとは異なるアプローチによるシーケンシング技術は、2008年から2009年にかけて初めて「第三世代」として説明された。[4] 現在、第三世代シーケンシング技術開発の中心となっている企業は、Pacific Biosciences、Oxford Nanopore Technology、Quantapore(カリフォルニア州)、Stratos(ワシントン州)などである。これらの企業は、単一DNA分子のシーケ ンシングに対して、根本的に異なるアプローチを取っている。 PacBioはゼロモード導波路の特性を基に、単分子リアルタイムシーケンシング(SMRT)のシーケンシングプラットフォームを開発した。シグナルは、zLウェルの底に結合したDNAポリメラーゼによって組み込まれた各ヌクレオチドからの蛍光発光の形をとる。 オックスフォード・ナノポア社の技術は、DNA分子をナノスケールの孔構造に通し、孔の周囲の電界の変化を測定するものである。一方、クオンタペーは独自 のナノポアアプローチを採用している。ストラトス・ジェノミクス社は、ナノポアssDNA読み取りのシグナル対ノイズの課題を回避するために、DNA塩基 をポリマー挿入物「Xpandomers」で間隔を空けて配置している。 また、Helicosの単分子蛍光アプローチも注目に値するが、同社は2015年秋に破産した。 |
| Advantages Longer reads In comparison to the current generation of sequencing technologies, third generation sequencing has the obvious advantage of producing much longer reads. It is expected that these longer read lengths will alleviate numerous computational challenges surrounding genome assembly, transcript reconstruction, and metagenomics among other important areas of modern biology and medicine.[1] It is well known that eukaryotic genomes including primates and humans are complex and have large numbers of long repeated regions. Short reads from second generation sequencing must resort to approximative strategies in order to infer sequences over long ranges for assembly and genetic variant calling. Pair end reads have been leveraged by second generation sequencing to combat these limitations. However, exact fragment lengths of pair ends are often unknown and must also be approximated as well. By making long reads lengths possible, third generation sequencing technologies have clear advantages. Epigenetics Epigenetic markers are stable and potentially heritable modifications to the DNA molecule that are not in its sequence. An example is DNA methylation at CpG sites, which has been found to influence gene expression. Histone modifications are another example. The current generation of sequencing technologies rely on laboratory techniques such as ChIP-sequencing for the detection of epigenetic markers. These techniques involve tagging the DNA strand, breaking and filtering fragments that contain markers, followed by sequencing. Third generation sequencing may enable direct detection of these markers due to their distinctive signal from the other four nucleotide bases.[5] Portability and speed  MinION Portable Gene Sequencer, Oxford Nanopore Technologies Other important advantages of third generation sequencing technologies include portability and sequencing speed.[6] Since minimal sample preprocessing is required in comparison to second generation sequencing, smaller equipments could be designed. Oxford Nanopore Technology has recently commercialized the MinION sequencer. This sequencing machine is roughly the size of a regular USB flash drive and can be used readily by connecting to a laptop. In addition, since the sequencing process is not parallelized across regions of the genome, data could be collected and analyzed in real time. These advantages of third generation sequencing may be well-suited in hospital settings where quick and on-site data collection and analysis is demanded. |
利点 より長いリード 現在のシーケンシング技術と比較すると、第3世代シーケンシング技術には、より長いリードを生成できるという明白な利点がある。これらのより長いリード長 により、ゲノムアセンブリ、転写産物の再構成、メタゲノム解析など、現代の生物学や医学の重要な分野における数多くの計算上の課題が軽減されることが期待 されている。 霊長類やヒトを含む真核生物のゲノムは複雑であり、多数の長い繰り返し領域を持つことはよく知られている。第2世代シーケンサーによる短いリードでは、ア センブリや遺伝子変異の検出のために長距離の配列を推定するには近似的な戦略に頼らざるを得ない。これらの制限に対処するために、第2世代シーケンサーで はペアエンドリードが活用されている。しかし、ペアエンドの断片の長さが正確にわからないことが多く、これも近似的に推定しなければならない。長距離リー ドの長さを可能にする第3世代シーケンサー技術には、明確な利点がある。 エピジェネティクス エピジェネティックマーカーは、DNA分子の配列には存在しない、安定した潜在的に遺伝可能な修飾である。その例としては、遺伝子発現に影響を与えること が分かっているCpG部位におけるDNAメチル化がある。ヒストン修飾もその例である。現在のシーケンシング技術では、エピジェネティックマーカーの検出 にChIP-sequencingなどの実験室技術に依存している。これらの技術では、DNA鎖にタグ付けを行い、マーカーを含む断片を切断およびフィル タリングし、その後シーケンスを行う。第3世代のシーケンシングでは、他の4つのヌクレオチド塩基とは異なるこれらのマーカーのシグナルにより、これらの マーカーを直接検出できる可能性がある。[5] 可搬性とスピード MinIONポータブル遺伝子シーケンサー、オックスフォード・ナノポア・テクノロジー 第3世代シーケンシング技術のその他の重要な利点として、可搬性とシーケンシングのスピードが挙げられる。[6] 第2世代シーケンシングと比較して、サンプルの前処理が最小限で済むため、より小型の機器を設計できる。オックスフォード・ナノポア・テクノロジーは最 近、MinION シーケンサーを商品化した。このシーケンサーは通常のUSBフラッシュドライブとほぼ同じ大きさで、ノートパソコンに接続するだけで使用できる。さらに、 シーケンシングプロセスはゲノムの領域ごとに並列化されていないため、データをリアルタイムで収集し、分析することができる。このような第3世代シーケン サーの利点は、迅速なデータ収集と分析が求められる病院の現場に適しているかもしれない。 |
| Challenges Parts of this article (those related to long-read sequencing technologies producing low-accuracy reads. While true 5 years ago, circular consensus reads with the PacBio Sequel II long-read sequencer can easily achieve an even higher read accuracy than hybrid genome assembly with a combination of other sequencers. [1] PMID 31885515, 28364362, 31406327, 31897449, 31483244) need to be updated. Please help update this article to reflect recent events or newly available information. (January 2020) Third generation sequencing, as of 2008, faced important challenges mainly surrounding accurate identification of nucleotide bases; error rates were still much higher compared to second generation sequencing.[2] This is generally due to instability of the molecular machinery involved. For example, in PacBio’s single molecular and real time sequencing technology, the DNA polymerase molecule becomes increasingly damaged as the sequencing process occurs.[2] Additionally, since the process happens quickly, the signals given off by individual bases may be blurred by signals from neighbouring bases. This poses a new computational challenge for deciphering the signals and consequently inferring the sequence. Methods such as Hidden Markov Models, for example, have been leveraged for this purpose with some success.[5] On average, different individuals of the human population share about 99.9% of their genes. In other words, approximately only one out of every thousand bases would differ between any two person. The high error rates involved with third generation sequencing are inevitably problematic for the purpose of characterizing individual differences that exist between members of the same species.[citation needed] |
課題 この記事の一部(低精度リードを生成する長鎖シーケンス技術に関連する部分。5年前は事実であったが、PacBio Sequel II長鎖シーケンサーによる環状コンセンサスリードは、他のシーケンサーを組み合わせたハイブリッドゲノムアセンブリよりもさらに高いリード精度を簡単に 達成できる。[1] PMID 31885515, 28364362, 31406327, 31897449, 31483244) は更新する必要がある。最近の出来事や新たに利用可能になった情報を反映させるため、この記事を更新するのを手伝ってください。 (2020年1月) 2008年時点での第3世代シーケンシングは、主にヌクレオチド塩基の正確な同定を巡る重要な課題に直面していた。エラー率は第2世代シーケンシングと比 較して依然としてはるかに高かった。[2] これは一般的に、関与する分子機械の不安定性によるものである。例えば、PacBioの単分子リアルタイムシーケンシング技術では、シーケンシングプロセ スが進むにつれ、DNAポリメラーゼ分子が損傷を受ける度合いが高くなる。[2] さらに、プロセスが迅速に進むため、個々の塩基から発せられるシグナルが、近隣の塩基からのシグナルによってぼやけてしまう可能性がある。このため、シグ ナルを解読し、結果として配列を推定するという新たな計算上の課題が生じる。例えば、隠れマルコフモデルなどの手法が、この目的のために活用され、ある程 度の成果を上げている。[5] 平均すると、ヒトの個体は約99.9%の遺伝子を共有している。言い換 えれば、2人のヒトの間では、約1000塩基対につき1つだけが異なることになる。第三世代シーケンサーのエラー率の高さは、同じ種に属する個体間の違い を特徴づけるという目的においては、避けられない問題である。[要出典] |
| Genome assembly Genome assembly is the reconstruction of whole genome DNA sequences. This is generally done with two fundamentally different approaches. Reference alignment When a reference genome is available, as one is in the case of human, newly sequenced reads could simply be aligned to the reference genome in order to characterize its properties. Such reference based assembly is quick and easy but has the disadvantage of “hiding" novel sequences and large copy number variants. In addition, reference genomes do not yet exist for most organisms. De novo assembly De novo assembly is the alternative genome assembly approach to reference alignment. It refers to the reconstruction of whole genome sequences entirely from raw sequence reads. This method would be chosen when there is no reference genome, when the species of the given organism is unknown as in metagenomics, or when there exist genetic variants of interest that may not be detected by reference genome alignment. Given the short reads produced by the current generation of sequencing technologies, de novo assembly is a major computational problem. It is normally approached by an iterative process of finding and connecting sequence reads with sensible overlaps. Various computational and statistical techniques, such as de bruijn graphs and overlap layout consensus graphs, have been leveraged to solve this problem. Nonetheless, due to the highly repetitive nature of eukaryotic genomes, accurate and complete reconstruction of genome sequences in de novo assembly remains challenging. Pair end reads have been posed as a possible solution, though exact fragment lengths are often unknown and must be approximated.[7] 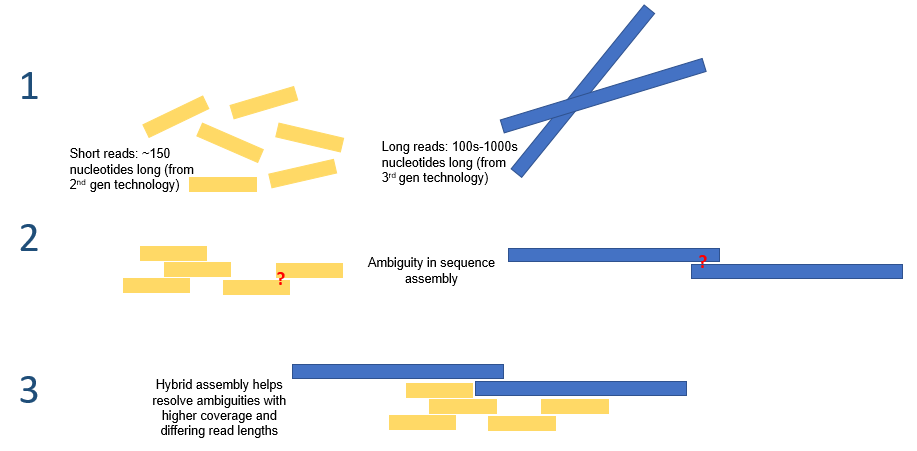 Hybrid assembly – the use of reads from 3rd gen sequencing platforms with shorts reads from 2nd gen platforms – may be used to resolve ambiguities that exist in genomes previously assembled using second generation sequencing. Short second generation reads have also been used to correct errors that exist in the long third generation reads. Hybrid assembly Long read lengths offered by third generation sequencing may alleviate many of the challenges currently faced by de novo genome assemblies. For example, if an entire repetitive region can be sequenced unambiguously in a single read, no computation inference would be required. Computational methods have been proposed to alleviate the issue of high error rates. For example, in one study, it was demonstrated that de novo assembly of a microbial genome using PacBio sequencing alone performed superior to that of second generation sequencing.[8] Third generation sequencing may also be used in conjunction with second generation sequencing. This approach is often referred to as hybrid sequencing. For example, long reads from third generation sequencing may be used to resolve ambiguities that exist in genomes previously assembled using second generation sequencing. On the other hand, short second generation reads have been used to correct errors in that exist in the long third generation reads. In general, this hybrid approach has been shown to improve de novo genome assemblies significantly.[9] |
ゲノムアセンブリ ゲノムアセンブリとは、ゲノムDNA配列全体の再構築を指す。これは一般的に、根本的に異なる2つのアプローチによって行われる。 参照アラインメント 参照ゲノムが利用可能な場合、ヒトの場合のように、新たに配列決定されたリードをその特性を明らかにするために参照ゲノムに単純にアラインメントすること ができる。このような参照ベースのアセンブリは迅速かつ容易であるが、新規配列やコピー数変異の「隠蔽」という欠点がある。さらに、ほとんどの生物には、 参照ゲノムはまだ存在していない。 デノボアセンブリ デノボアセンブリは、参照アラインメントの代替となるゲノムアセンブリのアプローチである。これは、生配列リードからゲノム配列全体を再構築することを指 す。この方法は、参照ゲノムが存在しない場合、メタゲノムのように生物種が不明である場合、または参照ゲノムアラインメントでは検出されない可能性がある 注目すべき遺伝的多型が存在する場合に選択される。 現行のシーケンシング技術で生成される短いリードを考慮すると、デノボアセンブリは大きな計算上の問題となる。通常、これは、意味のあるオーバーラップを 持つシーケンスリードを見つけ、つなぎ合わせる反復プロセスによって対処される。デ・ブルイーングラフやオーバーラップレイアウトコンセンサスグラフなど のさまざまな計算および統計的手法が、この問題の解決に活用されてきた。しかし、真核生物ゲノムの反復性の高さを考慮すると、デノボアセンブリによるゲノ ム配列の正確かつ完全な再構築は依然として困難である。ペアエンドリードは解決策のひとつとして考えられているが、正確な断片の長さは不明であることが多 く、近似値でなければならない。[7] ハイブリッドアセンブリ(第2世代プラットフォームのショートリードと第3世代プラットフォームのリードを併用する)は、第2世代シーケンシングでアセン ブルされたゲノムに存在するあいまいさを解決するために使用される可能性がある。また、第2世代シーケンシングのショートリードは、第3世代シーケンシン グのロングリードに存在するエラーを修正するために使用されることもある。 ハイブリッドアセンブリ 第3世代シーケンシングが提供する長いリード長は、現在デノボゲノムアセンブリが直面している多くの課題を軽減する可能性がある。例えば、反復領域全体が 1回のリードで明確にシーケンスできる場合、計算による推論は必要ない。高いエラー率の問題を軽減するための計算方法が提案されている。例えば、ある研究 では、PacBioシーケンシングのみを使用した微生物ゲノムのデノボアセンブリが、第2世代シーケンシングよりも優れていることが実証された。 第3世代シーケンサーは、第2世代シーケンサーと併用されることもある。このアプローチはハイブリッドシーケンシングと呼ばれることが多い。例えば、第3 世代シーケンサーによる長いリードは、第2世代シーケンサーで以前にアセンブルされたゲノムに存在する曖昧性を解決するために使用される。一方、第2世代 シーケンサーの短いリードは、第3世代シーケンサーの長いリードに存在するエラーを修正するために使用されている。一般的に、このハイブリッドアプローチ は、デノボゲノムアセンブリを大幅に改善することが示されている。[9] |
| Epigenetic markers DNA methylation (DNAm) – the covalent modification of DNA at CpG sites resulting in attached methyl groups – is the best understood component of epigenetic machinery. DNA modifications and resulting gene expression can vary across cell types, temporal development, with genetic ancestry, can change due to environmental stimuli and are heritable. After the discovery of DNAm, researchers have also found its correlation to diseases like cancer and autism.[10] In this disease etiology context DNAm is an important avenue of further research. Advantages The current most common methods for examining methylation state require an assay that fragments DNA before standard second generation sequencing on the Illumina platform. As a result of short read length, information regarding the longer patterns of methylation are lost.[5] Third generation sequencing technologies offer the capability for single molecule real-time sequencing of longer reads, and detection of DNA modification without the aforementioned assay.[11]  PacBio SMRT technology and Oxford Nanopore can use unaltered DNA to detect methylation. Oxford Nanopore Technologies’ MinION has been used to detect DNAm. As each DNA strand passes through a pore, it produces electrical signals which have been found to be sensitive to epigenetic changes in the nucleotides, and a hidden Markov model (HMM) was used to analyze MinION data to detect 5-methylcytosine (5mC) DNA modification.[5] The model was trained using synthetically methylated E. coli DNA and the resulting signals measured by the nanopore technology. Then the trained model was used to detect 5mC in MinION genomic reads from a human cell line which already had a reference methylome. The classifier has 82% accuracy in randomly sampled singleton sites, which increases to 95% when more stringent thresholds are applied.[5] Other methods address different types of DNA modifications using the MinION platform. Stoiber et al. examined 4-methylcytosine (4mC) and 6-methyladenine (6mA), along with 5mC, and also created software to directly visualize the raw MinION data in a human-friendly way.[12] Here they found that in E. coli, which has a known methylome, event windows of 5 base pairs long can be used to divide and statistically analyze the raw MinION electrical signals. A straightforward Mann-Whitney U test can detect modified portions of the E. coli sequence, as well as further split the modifications into 4mC, 6mA or 5mC regions.[12] It seems likely that in the future, MinION raw data will be used to detect many different epigenetic marks in DNA. PacBio sequencing has also been used to detect DNA methylation. In this platform, the pulse width – the width of a fluorescent light pulse – corresponds to a specific base. In 2010 it was shown that the interpulse distance in control and methylated samples are different, and there is a "signature" pulse width for each methylation type.[11] In 2012 using the PacBio platform the binding sites of DNA methyltransferases were characterized.[13] The detection of N6-methylation in C Elegans was shown in 2015.[14] DNA methylation on N6-adenine using the PacBio platform in mouse embryonic stem cells was shown in 2016.[15] Other forms of DNA modifications – from heavy metals, oxidation, or UV damage – are also possible avenues of research using Oxford Nanopore and PacBio third generation sequencing. Drawbacks Processing of the raw data – such as normalization to the median signal – was needed on MinION raw data, reducing real-time capability of the technology.[12] Consistency of the electrical signals is still an issue, making it difficult to accurately call a nucleotide. MinION has low throughput; since multiple overlapping reads are hard to obtain, this further leads to accuracy problems of downstream DNA modification detection. Both the hidden Markov model and statistical methods used with MinION raw data require repeated observations of DNA modifications for detection, meaning that individual modified nucleotides need to be consistently present in multiple copies of the genome, e.g. in multiple cells or plasmids in the sample. For the PacBio platform, too, depending on what methylation you expect to find, coverage needs can vary. As of March 2017, other epigenetic factors like histone modifications have not been discoverable using third-generation technologies. Longer patterns of methylation are often lost because smaller contigs still need to be assembled. |
エピジェネティックマーカー DNAメチル化(DNAm)は、CpG部位における共有結合によるDNAの修飾であり、メチル基の付着を伴う。これは、エピジェネティック機構の最もよく 理解されている要素である。DNAの修飾とそれによる遺伝子発現は、細胞の種類や時間的発達、遺伝的背景によって異なり、環境刺激によって変化し、遺伝性 がある。DNAmの発見後、研究者らは癌や自閉症などの疾患との相関関係も発見している。[10] この疾患の病因という観点では、DNAmはさらなる研究の重要な道筋となる。 利点 現在のメチル化状態を調べる最も一般的な方法では、イルミナプラットフォームの標準的な第2世代シーケンシングの前に、DNAを断片化するアッセイが必要 である。短いリード長により、より長いメチル化パターンに関する情報が失われる。第3世代シーケンシング技術は、前述のアッセイを必要とせずに、より長い リードの単分子リアルタイムシーケンシングとDNA修飾の検出を行うことができる。 PacBio SMRT技術とOxford Nanoporeは、修飾されていないDNAを用いてメチル化を検出することができる。 Oxford Nanopore TechnologiesのMinIONは、DNAmの検出に使用されている。DNA鎖が孔を通過する際に電気信号が発生し、その信号はヌクレオチドのエ ピジェネティックな変化に敏感であることが分かっている。隠れマルコフモデル(HMM)を使用してMinIONデータを分析し、5-メチルシトシン (5mC)DNA修飾を検出した。[5] このモデルは、合成的にメチル化された大腸菌DNAとナノポア技術で測定した結果の信号を使用して訓練された。その後、この学習済みモデルを使用して、参 照メチルームがすでに存在するヒト細胞株の MinION ゲノムリードから 5mC を検出した。この分類器は、ランダムに抽出された単一サイトにおいて 82% の精度を示し、より厳格な閾値を適用すると 95% にまで向上した。[5] MinION プラットフォームを使用して、他の手法では異なるタイプの DNA 修飾に対応している。Stoiber 氏らは、5mC とともに 4-メチルシトシン(4mC)と 6-メチルアデニン(6mA)を調べ、MinION の生データを直接、人間にとってわかりやすい形で視覚化するソフトウェアも作成した。[12] ここでは、メチル化状態が既知である大腸菌において、5 塩基対のイベントウィンドウを使用して MinION の生電気信号を分割し、統計的に分析できることがわかった。単純なマン・ホイットニーのU検定によって、大腸菌の配列の修飾部分を検出できるだけでなく、 さらに修飾部分を4mC、6mA、または5mC領域に分割することもできる。[12] 今後、MinIONの生データが、DNAのさまざまなエピジェネティックなマーカーを検出するために使用される可能性が高い。 PacBioシーケンシングもまた、DNAメチル化の検出に使用されている。このプラットフォームでは、蛍光パルスの幅であるパルス幅が特定の塩基に対応 する。2010年には、コントロールサンプルとメチル化サンプルにおけるパルス間の距離が異なり、各メチル化タイプに「シグネチャー」パルス幅があること が示された。[11] 2012年には、PacBioプラットフォームを使用して、DNAメチルトランスフェラーゼの結合部位が 特性が明らかにされた。[13] 2015年には、CエレガンスにおけるN6-メチル化の検出が示された。[14] 2016年には、マウス胚性幹細胞におけるPacBioプラットフォームを用いたN6-アデニン上のDNAメチル化が示された。[15] 重金属、酸化、または紫外線による損傷など、他のDNA修飾の形態も、Oxford NanoporeとPacBioの第3世代シーケンサーを使用した研究の対象となり得る。 欠点 MinIONの生データでは、中央値信号への正規化などの生データの処理が必要であり、技術のリアルタイム能力が低下していた。[12] 電気信号の一貫性は依然として問題であり、ヌクレオチドを正確に特定することが難しい。MinIONはスループットが低く、複数のオーバーラップするリー ドを取得することが困難であるため、下流のDNA修飾検出の精度に問題が生じる。MinIONの生データで使用される隠れマルコフモデルと統計的手法は、 検出のためにDNA修飾の繰り返し観察を必要とするため、個々の修飾ヌクレオチドは、サンプル内の複数の細胞またはプラスミドなど、ゲノムの複数のコピー に一貫して存在する必要がある。 PacBioプラットフォームの場合も、予想されるメチル化の種類によって、カバレッジのニーズは異なる。2017年3月現在、ヒストン修飾のような他の エピジェネティック因子は、第3世代の技術では検出できない。より長いメチル化パターンは、より小さなコンティグをアセンブルする必要があるため、しばし ば失われる。 |
| Transcriptomics Transcriptomics is the study of the transcriptome, usually by characterizing the relative abundances of messenger RNA molecules in the tissue under study. According to the central dogma of molecular biology, genetic information flows from double stranded DNA molecules to single stranded mRNA molecules where they can be readily translated into functional protein molecules. By studying the transcriptome, one can gain valuable insight into the regulation of gene expression. While expression levels can be more or less accurately depicted by second generation sequencing (we can assume that actual abundances of the population of transcripts are randomly sampled), transcript-level information still remains an important challenge.[16] As a consequence, the role of alternative splicing in molecular biology remains largely elusive. Third generation sequencing technologies hold promising prospects in resolving this issue by enabling sequencing of mRNA molecules at their full lengths. Alternative splicing Alternative splicing (AS) is the process by which a single gene may give rise to multiple distinct mRNA transcripts and consequently different protein translations.[17] Some evidence suggests that AS is a ubiquitous phenomenon and may play a key role in determining the phenotypes of organisms, especially in complex eukaryotes; all eukaryotes contain genes consisting of introns that may undergo AS. In particular, it has been estimated that AS occurs in 95% of all human multi-exon genes.[18] AS has undeniable potential to influence myriad biological processes. Advancing knowledge in this area has critical implications for the study of biology in general. Transcript reconstruction The current generation of sequencing technologies produce only short reads, putting tremendous limitation on the ability to detect distinct transcripts; short reads must be reverse engineered into original transcripts that could have given rise to the resulting read observations.[19] This task is further complicated by the highly variable expression levels across transcripts, and consequently variable read coverages across the sequence of the gene.[19] In addition, exons may be shared among individual transcripts, rendering unambiguous inferences essentially impossible.[17] Existing computational methods make inferences based on the accumulation of short reads at various sequence locations often by making simplifying assumptions.[19] Cufflinks takes a parsimonious approach, seeking to explain all the reads with the fewest possible number of transcripts.[20] On the other hand, StringTie attempts to simultaneously estimate transcript abundances while assembling the reads.[19] These methods, while reasonable, may not always identify real transcripts. A study published in 2008 surveyed 25 different existing transcript reconstruction protocols.[16] Its evidence suggested that existing methods are generally weak in assembling transcripts, though the ability to detect individual exons are relatively intact.[16] According to the estimates, average sensitivity to detect exons across the 25 protocols is 80% for Caenorhabditis elegans genes.[16] In comparison, transcript identification sensitivity decreases to 65%. For human, the study reported an exon detection sensitivity averaging to 69% and transcript detection sensitivity had an average of a mere 33%.[16] In other words, for human, existing methods are able to identify less than half of all existing transcript. Third generation sequencing technologies have demonstrated promising prospects in solving the problem of transcript detection as well as mRNA abundance estimation at the level of transcripts. While error rates remain high, third generation sequencing technologies have the capability to produce much longer read lengths.[21] Pacific Bioscience has introduced the iso-seq platform, proposing to sequence mRNA molecules at their full lengths.[21] It is anticipated that Oxford Nanopore will put forth similar technologies. The trouble with higher error rates may be alleviated by supplementary high quality short reads. This approach has been previously tested and reported to reduce the error rate by more than 3 folds.[22] |
トランスクリプトミクス トランスクリプトミクスは、通常、研究対象の組織におけるメッセンジャーRNA分子の相対的豊富度を特徴付けることによって、トランスクリプトームを研究 するものである。分子生物学の中心ドグマによると、遺伝情報は二重らせんDNA分子から単一鎖mRNA分子へと流れ、そこで容易に機能性タンパク質分子へ と翻訳される。トランスクリプトームを研究することによって、遺伝子発現の制御に関する貴重な洞察を得ることができる。 第二世代シーケンシングでは、発現レベルをある程度正確に描写することができる(転写産物の集団の実際の存在量がランダムにサンプリングされていると仮定 できる)が、転写レベルの情報は依然として重要な課題である。[16] その結果、分子生物学における選択的スプライシングの役割は依然としてほとんど解明されていない。第三世代シーケンシング技術は、mRNA分子の全長シー ケンシングを可能にすることで、この問題の解決に有望な見通しをもたらしている。 選択的スプライシング 選択的スプライシング(AS)とは、1つの遺伝子が複数の異なるmRNA転写物を生み出し、その結果、異なるタンパク質翻訳が起こるプロセスである。 [17] いくつかの証拠から、ASは普遍的な現象であり、特に複雑な真核生物において生物の表現型を決定する上で重要な役割を果たしている可能性があることが示唆 されている。すべての真核生物は、ASを受ける可能性があるイントロンから構成される遺伝子を含んでいる。特に、ヒトの多エクソン遺伝子の95%でASが 起こっていると推定されている。[18] ASは、数多くの生物学的プロセスに影響を及ぼす可能性を否定できない。この分野の知識の進歩は、生物学全般の研究に重大な影響を及ぼす。 転写産物の再構成 現行のシーケンシング技術では短いリードしか生成されないため、個々の転写産物を検出する能力には大きな限界がある。短いリードは、結果として得られた リードの観察結果を生み出した可能性のある元の転写産物にリバースエンジニアリングしなければならない。[19] この作業は、転写産物間の発現レベルが大きく異なるため、さらに複雑になる。その結果、遺伝子の配列全体にわたってリードのカバレッジが異なる。[19] さらに、エクソンは個々の転写産物間で共有されている可能性があり、明確な推論は基本的に不可能である。[ 17] 既存の計算方法は、さまざまな配列位置における短いリードの蓄積に基づいて推論を行うが、その際、単純化された仮定を置くことが多い。[19] Cufflinksは、最小限の転写産物で全てのリードを説明しようとする、倹約的なアプローチを取る。[20] 一方、StringTieは、リードをアセンブルしながら、転写産物の存在量を同時に推定しようとする。[19] これらの方法は妥当ではあるが、常に実際の転写産物を特定できるとは限らない。 2008年に発表された研究では、25種類の異なる既存の転写産物再構成プロトコルが調査された。[16] その証拠によると、既存の方法は転写産物のアセンブリには概して弱いものの、個々のエクソンの検出能力は比較的損なわれていないことが示唆された。 [16] 推定によると、25種類のプロトコル全体におけるエクソン検出の平均感度は、線虫の遺伝子では80%であった。[16] これに対し、転写産物同定の感度は65%に減少する。ヒトの場合、この研究ではエクソン検出感度が平均69%、トランスクリプト検出感度はわずか33%と いう結果であった。[16] 言い換えれば、ヒトの場合、既存の方法では全トランスクリプトの半分以下しか同定できないということである。 第三世代シーケンシング技術は、転写産物の検出および転写産物のレベルでのmRNAの存在量の推定という問題の解決に有望な見通しを示している。エラー率 は依然として高いものの、第三世代シーケンシング技術は、はるかに長いリード長を生成する能力がある。[21] パシフィック・バイオサイエンス社は、iso-seqプラットフォームを導入し、mRNA分子を全長にわたってシーケンスすることを提案している。 [21] オックスフォード・ナノポア社も同様の技術を開発する見込みである。エラー率の高さという問題は、高品質の短いリードを補足することで緩和できる可能性が ある。このアプローチは以前にもテストされ、エラー率を3分の1以下に削減できることが報告されている。[22] |
| Metagenomics Metagenomics is the analysis of genetic material recovered directly from environmental samples. Advantages The main advantage for third-generation sequencing technologies in metagenomics is their speed of sequencing in comparison to second generation techniques. Speed of sequencing is important for example in the clinical setting (i.e. pathogen identification), to allow for efficient diagnosis and timely clinical actions. Oxford Nanopore's MinION was used in 2015 for real-time metagenomic detection of pathogens in complex, high-background clinical samples. The first Ebola virus (EBOV) read was sequenced 44 seconds after data acquisition.[23] There was uniform mapping of reads to genome; at least one read mapped to >88% of the genome. The relatively long reads allowed for sequencing of a near-complete viral genome to high accuracy (97–99% identity) directly from a primary clinical sample.[23] A common phylogenetic marker for microbial community diversity studies is the 16S ribosomal RNA gene. Both MinION and PacBio's SMRT platform have been used to sequence this gene.[24][25] In this context the PacBio error rate was comparable to that of shorter reads from 454 and Illumina's MiSeq sequencing platforms.[citation needed] Drawbacks MinION's high error rate (~10-40%) prevented identification of antimicrobial resistance markers, for which single nucleotide resolution is necessary. For the same reason, eukaryotic pathogens were not identified.[23] Ease of carryover contamination when re-using the same flow cell (standard wash protocols don’t work) is also a concern. Unique barcodes may allow for more multiplexing. Furthermore, performing accurate species identification for bacteria, fungi and parasites is very difficult, as they share a larger portion of the genome, and some only differ by <5%. The per base sequencing cost is still significantly more than that of MiSeq. However, the prospect of supplementing reference databases with full-length sequences from organisms below the limit of detection from the Sanger approach;[24] this could possibly greatly help the identification of organisms in metagenomics. |
メタゲノミクス メタゲノミクスとは、環境サンプルから直接回収した遺伝物質の解析である。 利点 メタゲノムにおける第3世代シーケンシング技術の主な利点は、第2世代技術と比較したシーケンスのスピードである。シーケンスのスピードは、例えば臨床現場(すなわち病原体の特定)において、効率的な診断と迅速な臨床対応を可能にするために重要である。 オックスフォード・ナノポアのMinIONは、2015年に複雑でバックグラウンドが高い臨床サンプル中の病原体のリアルタイムメタゲノム検出に使用され た。最初のエボラウイルス(EBOV)のリードは、データ取得から44秒後に配列決定された。[23] リードはゲノムに均一にマッピングされ、少なくとも1つのリードはゲノムの88%以上にマッピングされた。比較的長いリードにより、一次臨床サンプルから 直接、高い精度(同一性97~99%)でほぼ完全なウイルスゲノムのシーケンスが可能となった。 微生物群の多様性研究における一般的な系統発生マーカーは、16SリボソームRNA遺伝子である。MinIONとPacBioのSMRTプラットフォーム の両方が、この遺伝子の配列決定に使用されている。[24][25]この文脈において、PacBioのエラー率は、454およびIlluminaの MiSeqシーケンシングプラットフォームからの短いリードのエラー率と同等であった。[要出典] 欠点 MinIONのエラー率が高い(~10~40%)ため、一塩基分解能が必要な薬剤耐性マーカーの特定が妨げられた。同じ理由で、真核病原体の特定もできな かった。[23] 同じフローセルを再使用する際のキャリーオーバー汚染のしやすさ(標準的な洗浄プロトコルでは対応できない)も懸念事項である。固有のバーコードにより、 多重化の可能性が高まる可能性がある。さらに、細菌、真菌、寄生虫の正確な種同定を行うのは非常に困難である。これらの生物はゲノムの大部分を共有してお り、一部はわずか5%しか異ならないからだ。 塩基配列決定コストは、MiSeqよりも依然としてかなり高い。しかし、サンガー法では検出限界以下の生物の全長配列をリファレンスデータベースに追加するという見通しがある。[24] これは、メタゲノムにおける生物の同定に大いに役立つ可能性がある。 |
| First-generation sequencing Second-generation sequencing |
第一世代シーケンサー 第二世代シーケンサー |
++
| DNA(デオキシリボ核酸)は、その構造内に
生命を創造し維持する生命システム全てに影響を及ぼす分子装置であるタンパク質やノンコーディングRNAの構築に必要なコードをもつ生命の青写真ともいえ
るものです。DNA配列を理解することで、研究者がRNAだけでなくタンパク質の構造や機能を解明することが可能となり、疾患の根底にある原因を理解する
ことが可能となってきました。次世代シーケンシング(NGS)は数千から数百万もの
DNA分子を同時に配列決定可能な強力な基盤技術です。次世代シーケンシングでは、複数個体を同時に配列決定できるなど高度かつ高速な処理が可能であるこ
とから、個の医療、遺伝性疾患、および臨床診断学といった分野に変革をもたらしています。 https://www.cosmobio.co.jp/support/technology/a/next-generation-sequencing-introduction-apb.asp |
 An Illumina HiSeq 2000 sequencing machine |
| 【旧世代のシークエンシンング】 サンガーシーケンシングでは高精度のDNA依存性ポリメラーゼを利用し、単一鎖DNA鋳型に対して相補的なコピーを産生します(1, 2, 3)。各反応において、鋳型に相補的なひとつのプライマーがその3'末端よりDNA合成反応を開始します。デオキシヌクレオチドまたは核酸はDNA単量体 ですが、これが次々に鋳型依存的に加えられ、伸展しているプライマー末端の3'ヒドロキシルと取り入れられてくる核酸の5'三リン酸塩基との間にリン酸ジ エステル結合が形成されます(図.1)(1)。 各反応にはそれぞれ各DNA塩基(つまり、A, G, T, およびC)に対応する4種のジデオキシヌクレオチドの混合物が含まれています。これらのジデオキシヌクレオチドはDNA単量体と類似しており伸展鎖に組込 まれますが、天然のデオキシヌクレオチドとは以下の2つの点が異なります。 さらなるDNA伸展に必要な3'ヒドロキシル基が欠損しており、DNA分子に組込まれると鎖の産生が停止する。 各ジデオキシヌクレオチドには独自の蛍光染色が付加されており、DNA配列を自動的に検出できる(3, 4, 5)。 反応ごとに多数の異なる鎖長のDNA断片コピーが作製され、この伸展は鋳型分子のヌクレオチド位の何れかにおいて対応するジデオキシヌクレオチドにより終 止されます(図.1)。反応混合液をシーケンシング機器にロードする際は、手動で平板ゲル上にロードするか毛細管を用いて自動でロードし、電気泳動を行っ てDNA分子を大きさで分類します。DNA配列はジデオキシヌクレオチドがゲルを通過する際にその蛍光発光を読み取ります(図.2)(5)。近年のサン ガーシーケンシング機器はキャピラリーを基盤とした自動化電気泳動を利用しており、通常、8~96シーケンシング反応を同時に分析することができます。 https://www.cosmobio.co.jp/support/technology/a/next-generation-sequencing-introduction-apb.asp |
★前史 「1975年、サンガーとアラン・コールソンは、DNAポリメラーゼと放射性標識ヌクレオチドを用いた塩基配列決定法を発表し、これをプラス・マイナス法 と呼んだ[21][22]。これらはポリアクリルアミドゲル上の電気泳動(ポリアクリルアミドゲル電気泳動と呼ばれる)によって分画され、オートラジオグ ラフィーを用いて可視化された。この方法では一度に80塩基までの塩基配列を決定することができ、大きな進歩であったが、それでも非常に手間のかかるもの であった。それでも1977年、彼のグループは一本鎖のバクテリオファージφX174の5,386ヌクレオチドのほとんどを配列決定することができ、初め てDNAベースのゲノムを完全に配列決定した。プラス・マイナス法を改良した結果、連鎖終結法、すなわちサンガー法(下記参照)が生まれ、その後の四半世 紀にわたる研究で最も広く使われたDNA配列決定、ゲノムマッピング、データ保存、バイオインフォマティクス解析の技術の基礎となった。[25]同年、 ハーバード大学のウォルター・ギルバートとアラン・マクサムは、既知の塩基におけるDNAの優先的切断を伴う、DNA配列決定のマクサム・ギルバート法 (化学的方法としても知られる)を独自に開発したが、これはあまり効率的な方法ではなかった[26][27]。核酸の配列決定における画期的な業績によ り、ギルバートとサンガーは、ポール・バーグ(組み換えDNA)と共に1980年のノーベル化学賞の半分を受賞した」(→「ゲノミクス」) |
| Massive parallel sequencing or
massively parallel sequencing is any of several high-throughput
approaches to DNA sequencing using the concept of massively parallel
processing; it is also called next-generation sequencing (NGS) or
second-generation sequencing. Some of these technologies emerged
between 1993 and 1998 [1][2][3][4][5] and have been commercially
available since 2005. These technologies use miniaturized and
parallelized platforms for sequencing of 1 million to 43 billion short
reads (50 to 400 bases each) per instrument run. Many NGS platforms differ in engineering configurations and sequencing chemistry. They share the technical paradigm of massive parallel sequencing via spatially separated, clonally amplified DNA templates or single DNA molecules in a flow cell. This design is very different from that of Sanger sequencing—also known as capillary sequencing or first-generation sequencing—which is based on electrophoretic separation of chain-termination products produced in individual sequencing reactions.[6] This methodology allows sequencing to be completed on a larger scale.[7] |
大規模並列シーケンシングまたは大規模並列シーケンシングは、大規模並
列処理の概念を用いたDNAシーケンシングのいくつかの高処理アプローチのうちの1つである。次世代シーケンシング(NGS)または第2世代シーケンシン
グとも呼ばれる。これらの技術の一部は1993年から1998年の間に登場し[1][2][3][4][5]、2005年以降は商業的に利用可能となって
いる。これらの技術では、小型化および並列化されたプラットフォームを使用し、1回の実行につき100万から430億の短いリード(各50から400塩
基)のシーケンスを行う。 多くのNGSプラットフォームは、エンジニアリング構成やシーケンス化学が異なる。それらは、フローセル内の空間的に分離されたクローン増幅DNAテンプ レートまたは単一DNA分子を介した大規模並列シーケンスという技術的パラダイムを共有している。この設計は、個々のシーケンス反応で生成された鎖停止産 物の電気泳動分離に基づくサンガーシーケンス(キャピラリーシーケンスまたは第一世代シーケンスとも呼ばれる)の設計とは大きく異なる。[6] この方法論により、より大規模なシーケンスが可能になる。[7] |
| Maxam–Gilbert
sequencing Third-generation sequencing |
|
| NGS platforms DNA sequencing with commercially available NGS platforms is generally conducted with the following steps. First, DNA sequencing libraries are generated by clonal amplification by PCR in vitro. Second, the DNA is sequenced by synthesis, such that the DNA sequence is determined by the addition of nucleotides to the complementary strand rather than through chain-termination chemistry. Third, the spatially segregated, amplified DNA templates are sequenced simultaneously in a massively parallel fashion without the requirement for a physical separation step. These steps are followed in most NGS platforms, but each utilizes a different strategy.[8] NGS parallelization of the sequencing reactions generates hundreds of megabases to gigabases of nucleotide sequence reads in a single instrument run. This has enabled a drastic increase in available sequence data and fundamentally changed genome sequencing approaches in the biomedical sciences.[9] Newly emerging NGS technologies and instruments have further contributed to a significant decrease in the cost of sequencing nearing the mark of $1000 per genome sequencing.[10][11] As of 2014, massively parallel sequencing platforms are commercially available and their features are summarized in the table. As the pace of NGS technologies is advancing rapidly, technical specifications and pricing are in flux. 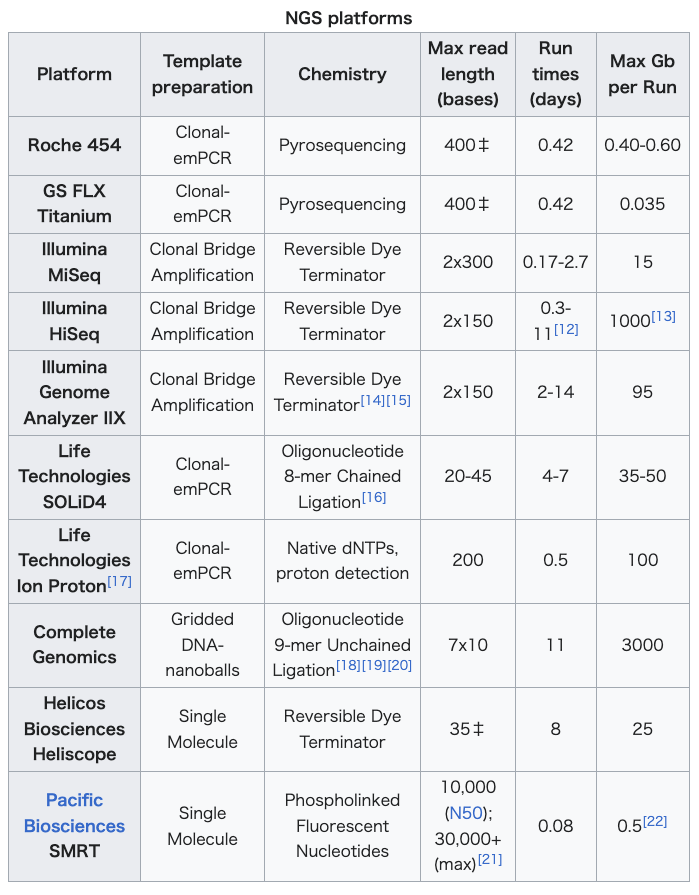 Run times and gigabase (Gb) output per run for single-end sequencing are noted. Run times and outputs approximately double when performing paired-end sequencing. ‡Average read lengths for the Roche 454 and Helicos Biosciences platforms.[23] |
NGSプラットフォーム 市販のNGSプラットフォームを用いたDNAシークエンシングは、一般的に以下の手順で行われる。まず、インビトロでのPCRによるクローン増幅により、 DNAシークエンシングライブラリが生成される。次に、DNAは合成によりシークエンシングされ、DNA配列は、鎖終結化学反応ではなく相補鎖へのヌクレ オチドの付加により決定される。第三に、空間的に分離された増幅されたDNAテンプレートは、物理的な分離ステップを必要とせずに、大規模な並列方式で同 時に配列決定される。これらのステップはほとんどのNGSプラットフォームで踏襲されているが、それぞれ異なる戦略が採用されている。 NGSのシーケンス反応の並列化により、1回の装置実行で数百メガベースからギガベースのヌクレオチド配列リードが生成される。これにより、利用可能な シーケンスデータの大幅な増加が可能になり、生物医学科学におけるゲノムシーケンスのアプローチが根本的に変化した。[9] 新たに登場したNGS技術と装置は、シーケンスコストの大幅な削減にさらに貢献し、ゲノムシーケンス1回あたり1000ドルという目標に近づいている。 [10][11] 2014年現在、超並列シーケンシングプラットフォームは市販されており、その特徴は表にまとめられている。NGS技術の進歩は急速であるため、技術仕様 や価格設定は流動的である。 シングルエンドシーケンスの実行時間と1回の実行ごとのギガベース(Gb)出力が記載されている。ペアエンドシーケンスを実行すると、実行時間と出力は約 2倍になる。‡ ロシュ454およびHelicosバイオサイエンスプラットフォームの平均リード長。[23] |
| Template preparation methods for NGS Two methods are used in preparing templates for NGS reactions: amplified templates originating from single DNA molecules, and single DNA molecule templates. For imaging systems which cannot detect single fluorescence events, amplification of DNA templates is required. The three most common amplification methods are emulsion PCR (emPCR), rolling circle and solid-phase amplification. The final distribution of templates can be spatially random or on a grid. Emulsion PCR In emulsion PCR methods, a DNA library is first generated through random fragmentation of genomic DNA. Single-stranded DNA fragments (templates) are attached to the surface of beads with adaptors or linkers, and one bead is attached to a single DNA fragment from the DNA library. The surface of the beads contains oligonucleotide probes with sequences that are complementary to the adaptors binding the DNA fragments. The beads are then compartmentalized into water-oil emulsion droplets. In the aqueous water-oil emulsion, each of the droplets capturing one bead is a PCR microreactor that produces amplified copies of the single DNA template.[24][25][26] Gridded rolling circle nanoballs Amplification of a population of single DNA molecules by rolling circle amplification in solution is followed by capture on a grid of spots sized to be smaller than the DNAs to be immobilized.[27][28][29][30] DNA colony generation (Bridge amplification) Forward and reverse primers are covalently attached at high-density to the slide in a flow cell. The ratio of the primers to the template on the support defines the surface density of the amplified clusters. The flow cell is exposed to reagents for polymerase-based extension, and priming occurs as the free/distal end of a ligated fragment "bridges" to a complementary oligo on the surface. Repeated denaturation and extension results in localized amplification of DNA fragments in millions of separate locations across the flow cell surface. Solid-phase amplification produces 100–200 million spatially separated template clusters, providing free ends to which a universal sequencing primer is then hybridized to initiate the sequencing reaction.[24][25] This technology was filed for a patent in 1997 from Glaxo-Welcome's Geneva Biomedical Research Institute (GBRI), by Pascal Mayer, Eric Kawashima, and Laurent Farinelli,[4][5] and was publicly presented for the first time in 1998.[31] In 1994 Chris Adams and Steve Kron filed a patent on a similar, but non-clonal, surface amplification method, named “bridge amplification”[3] adapted for clonal amplification in 1997 by Church and Mitra.[27][28] Single-molecule templates Protocols requiring DNA amplification are often cumbersome to implement and may introduce sequencing errors. The preparation of single-molecule templates is more straightforward and does not require PCR, which can introduce errors in the amplified templates. AT-rich and GC-rich target sequences often show amplification bias, which results in their underrepresentation in genome alignments and assemblies. Single molecule templates are usually immobilized on solid supports using one of at least three different approaches. In the first approach, spatially distributed individual primer molecules are covalently attached to the solid support. The template, which is prepared by randomly fragmenting the starting material into small sizes (for example,~200–250 bp) and adding common adapters to the fragment ends, is then hybridized to the immobilized primer. In the second approach, spatially distributed single-molecule templates are covalently attached to the solid support by priming and extending single-stranded, single-molecule templates from immobilized primers. A common primer is then hybridized to the template. In either approach, DNA polymerase can bind to the immobilized primed template configuration to initiate the NGS reaction. Both of the above approaches are used by Helicos BioSciences. In a third approach, spatially distributed single polymerase molecules are attached to the solid support, to which a primed template molecule is bound. This approach is used by Pacific Biosciences. Larger DNA molecules (up to tens of thousands of base pairs) can be used with this technique and, unlike the first two approaches, the third approach can be used with real-time methods, resulting in potentially longer read lengths. |
NGSのためのテンプレート準備方法 NGS反応用のテンプレートを準備するには、2つの方法が用いられる。1つは、単一DNA分子から増幅されたテンプレート、もう1つは、単一DNA分子テ ンプレートである。単一の蛍光事象を検出できないイメージングシステムでは、DNAテンプレートの増幅が必要である。最も一般的な増幅方法としては、エマ ルジョンPCR(emPCR)、ローリングサークル増幅、固相増幅の3つがある。テンプレートの最終的な配置は、空間的にランダムでもグリッド上でもよ い。 エマルジョン PCR エマルジョン PCR 法では、まずゲノム DNA をランダムに断片化して DNA ライブラリを生成する。一本鎖 DNA 断片(テンプレート)をアダプターまたはリンカーとともにビーズの表面に付着させ、DNA ライブラリから得た一本の DNA 断片に 1 つのビーズを付着させる。ビーズの表面には、DNA断片を結合するアダプターと相補的な配列を持つオリゴヌクレオチドプローブが結合している。その後、 ビーズは水相と油相のエマルション小滴に区画化される。水相と油相のエマルション中では、各小滴は1つのビーズを捕捉しており、これは1つのDNAテンプ レートを増幅したコピーを生成するPCRマイクロリアクターである。[24][25][26] グリッドローリングサークルナノボール 溶液中でのローリングサークル増幅による単一DNA分子の増幅は、固定化されるDNAよりも小さいサイズのスポットのグリッドへの捕捉が続く。[27][28][29][30] DNAコロニー生成(ブリッジ増幅 フォワードおよびリバースプライマーはフローセル内のスライドに高密度で共有結合する。支持体上のテンプレートに対するプライマーの比率は、増幅されたク ラスターの表面密度を決定する。フローセルは、ポリメラーゼをベースとする伸長反応用の試薬にさらされ、ライゲーションされた断片の遊離末端/遠位末端が 表面上の相補的オリゴに「橋渡し」されることでプライミングが行われる。変性と伸長を繰り返すことで、フローセル表面の数百万の別々の位置でDNA断片の 局所的な増幅が起こる。固相増幅により、1億から2億の空間的に分離したテンプレートクラスターが生成され、ユニバーサルシーケンスプライマーがハイブリ ダイズする遊離末端が提供される。これにより、シーケンス反応が開始される。[24][25] この技術は、1997年にグラクソ・ウェルカムのジュネーブ生物医学研究所(GBRI)のパスカル・メイヤー、エリック・カワシマ、ローラン・ファリネッ リにより特許出願された であり、1998年に初めて公に発表された。[31] 1994年、クリス・アダムスとスティーブ・クロンは、クローン増幅ではないが類似した表面増幅法である「ブリッジ増幅」[3]の特許を出願した。この方 法は、1997年にチャーチとミトラによってクローン増幅用に改良された。[27][28] 単一分子テンプレート DNA増幅を必要とするプロトコルは、実施するのが煩雑であることが多く、配列決定エラーを引き起こす可能性もある。単一分子テンプレートの準備はより簡 単であり、増幅テンプレートにエラーを引き起こす可能性のあるPCRを必要としない。ATに富む領域やGCに富む領域の標的配列は、増幅バイアスを示すこ とが多く、ゲノムのアラインメントやアセンブリにおいて過小評価される結果となる。通常、単一分子テンプレートは、少なくとも3つの異なるアプローチのう ちの1つを使用して、固体支持体に固定化される。最初の方法では、空間的に分散した個々のプライマー分子が固体支持体に共有結合で固定される。テンプレー トは、出発材料をランダムに断片化して小さなサイズ(例えば、200~250bp程度)にし、断片末端に共通アダプターを付加して調製され、その後、固定 化されたプライマーにハイブリダイズされる。2つ目のアプローチでは、空間的に分散した単一分子テンプレートが、固定化プライマーから単鎖の単一分子テン プレートをプライミングして伸長させることにより、固相支持体に共有結合で固定される。次に、共通プライマーがテンプレートにハイブリダイズする。いずれ のアプローチでも、DNAポリメラーゼは固定化されたプライミングされたテンプレート構造に結合し、NGS反応を開始することができる。上記の2つのアプ ローチは、Helicos BioSciences社によって使用されている。第3の手法では、空間的に分散した単一のポリメラーゼ分子が、プライミングされたテンプレート分子が結 合した固体支持体に結合される。この手法はパシフィック・バイオサイエンシズ社によって使用されている。この技術では、より大きなDNA分子(数万塩基対 まで)を使用することができ、最初の2つの手法とは異なり、第3の手法はリアルタイム法と併用できるため、より長いリード長を実現できる可能性がある。 |
| Sequencing approaches Sequencing by synthesis The objective for sequential sequencing by synthesis (SBS) is to determine the sequencing of a DNA sample by detecting the incorporation of a nucleotide by a DNA polymerase. An engineered polymerase is used to synthesize a copy of a single strand of DNA and the incorporation of each nucleotide is monitored. The principle of sequencing by synthesis was first described in 1993 [1] with improvements published some years later.[32] The key parts are highly similar for all embodiments of SBS and include (1) amplification of DNA to enhance the subsequent signal and to attach the DNA to be sequenced to a solid support, (2) generation of single stranded DNA on the solid support, (3) incorporation of nucleotides using an engineered polymerase and (4) detection of the incorporation of nucleotide. Then steps 3-4 are repeated and the sequence is assembled from the signals obtained in step 4. This principle of sequencing-by-synthesis has been used for almost all massive parallel sequencing instruments, including 454, PacBio, IonTorrent, Illumina and MGI. Pyrosequencing The principle of Pyrosequencing was first described in 1993 [1] by combining a solid support with an engineered DNA polymerase lacking 3´to 5´exonuclease activity (proof-reading) and luminescence real-time detection using the firefly luciferase. All the key concepts of sequencing by synthesis were introduced, including (1) amplification of DNA to enhance the subsequent signal and attach the DNA to be sequenced (template) to a solid support, (2) generation of single stranded DNA on the solid support (3) incorporation of nucleotides using an engineered polymerase and (4) detection of the incorporated nucleotide by light detection in real-time. In a follow-up article,[2] the concept was further developed and in 1998, an article [32] was published in which the authors showed that non-incorporated nucleotides could be removed with a fourth enzyme (apyrase) allowing sequencing by synthesis to be performed without the need for washing away non-incorporated nucleotides. Sequencing by reversible terminator chemistry This approach uses reversible terminator-bound dNTPs in a cyclic method that comprises nucleotide incorporation, fluorescence imaging and cleavage. A fluorescently-labeled terminator is imaged as each dNTP is added and then cleaved to allow incorporation of the next base. These nucleotides are chemically blocked such that each incorporation is a unique event. An imaging step follows each base incorporation step, then the blocked group is chemically removed to prepare each strand for the next incorporation by DNA polymerase. This series of steps continues for a specific number of cycles, as determined by user-defined instrument settings. The 3' blocking groups were originally conceived as either enzymatic[33] or chemical reversal[14][15] The chemical method has been the basis for the Solexa and Illumina machines. Sequencing by reversible terminator chemistry can be a four-colour cycle such as used by Illumina/Solexa, or a one-colour cycle such as used by Helicos BioSciences. Helicos BioSciences used “virtual Terminators”, which are unblocked terminators with a second nucleoside analogue that acts as an inhibitor. These terminators have the appropriate modifications for terminating or inhibiting groups so that DNA synthesis is terminated after a single base addition.[25][34][35] Sequencing-by-ligation mediated by ligase enzymes In this approach, the sequence extension reaction is not carried out by polymerases but rather by DNA ligase and either one-base-encoded probes or two-base-encoded probes. In its simplest form, a fluorescently labelled probe hybridizes to its complementary sequence adjacent to the primed template. DNA ligase is then added to join the dye-labelled probe to the primer. Non-ligated probes are washed away, followed by fluorescence imaging to determine the identity of the ligated probe. The cycle can be repeated either by using cleavable probes to remove the fluorescent dye and regenerate a 5′-PO4 group for subsequent ligation cycles (chained ligation[16][36]) or by removing and hybridizing a new primer to the template (unchained ligation[18][19]). Phospholinked Fluorescent Nucleotides or Real-time sequencing Pacific Biosciences is currently leading this method. The method of real-time sequencing involves imaging the continuous incorporation of dye-labelled nucleotides during DNA synthesis: single DNA polymerase molecules are attached to the bottom surface of individual zero-mode waveguide detectors (Zmw detectors) that can obtain sequence information while phospholinked nucleotides are being incorporated into the growing primer strand. Pacific Biosciences uses a unique DNA polymerase which better incorporates phospholinked nucleotides and enables the resequencing of closed circular templates. While single-read accuracy is 87%, consensus accuracy has been demonstrated at 99.999% with multi-kilobase read lengths.[37][38] In 2015, Pacific Biosciences released a new sequencing instrument called the Sequel System, which increases capacity approximately 6.5-fold.[39][40] |
シーケンスアプローチ 合成によるシーケンス 合成シーケンス(SBS)の目的は、DNAポリメラーゼによるヌクレオチドの取り込みを検出することで、DNAサンプルのシーケンスを決定することであ る。 設計されたポリメラーゼが、DNAの単鎖のコピーを合成するために使用され、各ヌクレオチドの取り込みがモニターされる。合成シーケンシングの原理は、 1993年に初めて発表され[1]、その後数年間に改良が加えられた。[32] SBSのすべての実施態様において、重要な部分は非常に類似しており、(1) その後のシグナルを増幅し、 シグナルを増幅し、配列決定するDNAを固体支持体に結合させる、(2) 固体支持体上で一本鎖DNAを生成する、(3) 改変ポリメラーゼを使用してヌクレオチドを取り込む、(4) ヌクレオチドの取り込みを検出する。その後、ステップ3から4が繰り返され、ステップ4で得られたシグナルから配列が組み立てられる。このシーケンス・バ イ・シンセシスの原理は、454、PacBio、IonTorrent、Illumina、MGIなど、ほぼすべての大量並列シーケンシング装置で使用さ れている。 パイロシークエンシング パイロシークエンシングの原理は、1993年に初めて発表された[1]。3´から5´エキソヌクレアーゼ活性(校正機能)を欠くように設計されたDNAポ リメラーゼと、ホタルルシフェラーゼを用いた発光リアルタイム検出を組み合わせたものである。合成によるシーケンシングのすべての主要な概念が紹介され た。これには、(1) その後のシグナルを増幅し、シーケンスするDNA(テンプレート)を固相に結合させるためのDNAの増幅、(2) 固相上での一本鎖DNAの生成、(3) 改良型ポリメラーゼによるヌクレオチドの取り込み、(4) 取り込まれたヌクレオチドのリアルタイムでの光検出による検出、が含まれる。その後の記事[2]では、このコンセプトがさらに発展し、1998年には、第 4の酵素(アピラーゼ)によって非取り込みヌクレオチドを除去できることが示された論文[32]が発表された。これにより、非取り込みヌクレオチドを洗い 流す必要なく合成によるシーケンスを行うことが可能になった。 可逆的ターミネーター化学によるシーケンス このアプローチでは、可逆的ターミネーターに結合したdNTPsを、ヌクレオチド取り込み、蛍光イメージング、切断からなる循環的な方法で使用する。各 dNTPが添加され、次に次の塩基の取り込みを可能にするために切断されると、蛍光標識されたターミネーターがイメージ化される。これらのヌクレオチド は、各取り込みが唯一のイベントとなるように化学的にブロックされる。各塩基取り込みステップの後にイメージングステップが続き、その後、ブロックされた グループが化学的に除去され、DNAポリメラーゼによる次の取り込みに備えて各鎖が準備される。この一連のステップは、ユーザー定義の機器設定によって決 定される特定のサイクル数だけ継続される。3'ブロック基は、当初は酵素[33]または化学的[14][15]のいずれかで解除する方法として考案され た。化学的方法は、SolexaやIlluminaの機械の基礎となっている。可逆的ターミネーター化学による配列決定は、 Illumina/Solexaで使用されているような4色のサイクル、またはHelicos BioSciencesで使用されているような1色のサイクルが可能である。Helicos BioSciencesは「仮想ターミネーター」を使用していた。これは阻害剤として働く2番目のヌクレオシドアナログを持つ、ブロックされていないター ミネーターである。これらのターミネーターは、DNA合成を1塩基の付加後に停止させるために、停止または阻害基に適切な修飾が施されている。[25] [34][35] リガーゼ酵素によるライゲーション媒介シーケンシング このアプローチでは、配列の伸長反応はポリメラーゼではなく、DNAリガーゼと1塩基プローブまたは2塩基プローブによって行われる。最も単純な形では、 蛍光標識プローブがプライミングされたテンプレートの隣接する相補的配列にハイブリダイズする。次にDNAリガーゼが添加され、色素標識プローブをプライ マーに結合させる。ライゲーションされていないプローブは洗浄により除去され、続いて蛍光イメージングによりライゲーションされたプローブの同一性を決定 する。このサイクルは、切断可能なプローブを使用して蛍光色素を除去し、次のライゲーションサイクルのための5′-リン酸基を再生させる(連鎖ライゲー ション[16][36])か、または新しいプライマーをテンプレートから除去し、ハイブリダイズさせる(非連鎖ライゲーション[18][19])ことによ り、繰り返すことができる。 リン酸結合蛍光ヌクレオチドまたはリアルタイムシークエンシング パシフィック・バイオサイエンシズ社は現在、この方法で業界をリードしている。リアルタイムシークエンシングの手法では、DNA合成中に色素標識ヌクレオ チドを連続的に取り込む様子を画像化する。個々のDNAポリメラーゼ分子は、ゼロモード波導検出器(Zmw検出器)の底面に付着しており、伸長するプライ マー鎖にリン酸結合ヌクレオチドが取り込まれる際に、配列情報を取得することができる。パシフィック・バイオサイエンシズ社は、リン酸結合ヌクレオチドを よりよく取り込む独自のDNAポリメラーゼを使用しており、閉環状テンプレートの再シークエンシングを可能にしている。シングルリードの精度は87%であ るが、マルチキロベースリード長ではコンセンサス精度が99.999%であることが実証されている。[37][38] 2015年、パシフィック・バイオサイエンシズは、シーケンス能力を約6.5倍に高めた新シーケンス装置「Sequelシステム」を発表した。[39] [40] |
| Clinical metagenomic sequencing First-generation sequencing (Sanger sequencing) Third-generation sequencing RNA Velocity |
臨床メタゲノムシーケンス 第一世代シーケンサー(Sanger sequencing) 第三世代シーケンサー RNAの速度 |
| Illumina, Inc.
is an American biotechnology company, headquartered in San Diego,
California. Incorporated on April 1, 1998, Illumina develops,
manufactures, and markets integrated systems for the analysis of
genetic variation and biological function. The company provides a line
of products and services that serves the sequencing, genotyping and
gene expression, and proteomics markets, and serves more than 155
countries.[2] Illumina's customers include genomic research centers, pharmaceutical companies, academic institutions, clinical research organizations, and biotechnology companies.[1] History  Czarnik, Stuelpnagel, and Chee at their Illumina office in the summer of 1998 Illumina was founded in April 1998 by David Walt, Larry Bock, John Stuelpnagel, Anthony Czarnik, and Mark Chee. While working with CW Group, a venture-capital firm, Bock and Stuelpnagel uncovered what would become Illumina's BeadArray technology[3] at Tufts University and negotiated an exclusive license to that technology. In 1999, Illumina acquired Spyder Instruments (founded by Michal Lebl, Richard Houghten, and Jutta Eichler) for their technology of high-throughput synthesis of oligonucleotides. Illumina completed its initial public offering in July 2000.[4] Illumina began offering single nucleotide polymorphism (SNP) genotyping services in 2001 and launched its first system, the Illumina BeadLab, in 2002, using GoldenGate Genotyping technology. Illumina currently offers microarray-based products and services for an expanding range of genetic analysis sequencing, including SNP genotyping, gene expression, and protein analysis. Illumina's technologies are used by a broad range of academic, government, pharmaceutical, biotechnology, and other leading institutions around the globe. On January 26, 2007, the company completed the acquisition of the British company Solexa, Inc. for ~$650M.[5] Solexa was founded in June 1998 by Shankar Balasubramanian and David Klenerman to develop and commercialize genome-sequencing technology invented by the founders at the University of Cambridge. Solexa, Inc. was formed in 2005 when Solexa Ltd reversed into Lynx Therapeutics of Hayward.[6] Illumina also uses the DNA colony sequencing technology, invented in 1997 by Pascal Mayer and Laurent Farinelli [7] and which was acquired by Solexa in 2004 from Manteia Predictive Medicine. It is being used to perform a range of analyses, including whole genome resequencing, gene-expression analysis, and small ribonucleic acid (sRNA) analysis. In June 2009, Illumina announced the launch of their own Personal Full Genome Sequencing Service at a depth of 30X.[8] Until 2010, Illumina sold only instruments that were labeled "for research use only"; in early 2010, Illumina obtained FDA approval for its BeadXpress system to be used in clinical tests.[9][10] This was part of the company's strategy at the time to open its own CLIA lab and begin offering clinical genetic testing itself.[11] Illumina acquired Epicentre Biotechnologies, based in Madison, Wisconsin, on January 11, 2011.[12] On January 25, 2012, Hoffmann-La Roche made an unsolicited bid to buy Illumina for $44.50 per share or about $5.7 billion.[13][14] Roche tried other tactics, including raising its offer (to $51.00, for about $6.8 billion).[15] Illumina rejected the offer,[16] and Roche abandoned the offer in April.[17] In 2014, the company announced a multimillion-dollar product, HiSeq X Ten.[18][19] In January 2014, Illumina already held 70% of the market for genome-sequencing machines.[20] Illumina machines accounted for more than 90% of all DNA data produced.[21] In 2020, the company invested in the acquisition of the pre-commercial firm Enancio, which had developed a DNA data compression algorithm specifically targeting Illumina data capable of reducing storage footprint by 80% (e.g. 50 GB compressed to 10 GB).[22] On July 5, 2016, Jay Flatley, who had been CEO since 1999, assumed the role of executive chairman of the board of directors. Francis deSouza, who had been president of the company since 2013, took on the additional role of CEO.[23] In late 2015, Illumina spun off the company Grail, focused on blood testing for cancer tumors in the bloodstream. In 2017 Grail had planned to raise $1 billion in its second round of financing, and received funding from Bill Gates and Jeff Bezos investing $100 million in series A funding, and with Illumina maintaining a 20% holding share in Grail.[24] Grail is working with a blood test trial with over 120,000 women during scheduled mammogram visits in the states of Minnesota and Wisconsin, as well as a partnership with the Mayo Clinic. Grail uses Illumina sequencing technology for tests.[25] Grail planned to roll out the tests by 2019.[26] In September 2020, Illumina announced a proposed cash and stock deal to acquire Grail for $8 billion.[27][28] In November 2018, Illumina proposed the acquisition of Pacific Biosciences for $8.00 per share or around $1.2 billion in total.[29][30] In December 2019, the Federal Trade Commission (FTC) sued to block the acquisition.[31] The proposed deal was abandoned on January 2, 2020, with Illumina paying Pacific a $98 million termination fee.[32] In March 2021, the FTC sued to block Illumina's $7.1 billion vertical merger with Grail.[33][34] In July 2021, the European Commission opened an in-depth investigation into the Grail acquisition by Illumina.[35] Against the orders of active investigations by both the US FTC and the EU European Commission, Illumina publicly announced it had completed its acquisition of Grail on August 18, 2021.[36] The FTC urged Illumina to "unwind" the merger shortly after,[37] and in October 2021, the European Commission ordered Illumina to keep Grail a separate company[38] and adopted interim measures to prevent harm to competition, or face penalty payments up to 5% of their average daily turnover and/or fines up to 10% of their annual worldwide turnover under Articles 15 and 14 of the EU Merger Regulation respectively.[35] In September 2022, a US administrative judge ruled against the FTC's efforts to prevent the acquisition on antitrust grounds.[39] In April 2023, the FTC ordered Grail to be divested by Illumina.[40] In July 2023, the European Commission imposed a €432 million ($476 million) penalty on Illumina for closing the Grail acquisition without EU approval.[41] In September 2022, Illumina launched NovaSeq X and NovaSeq X Plus.[42] The NovaSeq X Plus can sequence 20,000 genomes per year, compared to 7,500 per year of Illumina's previous machines and generate up to 16 Tb of data per run.[42] The series includes redeveloped reagents, dyes, and polymerases which can be shipped at ambient temperature.[43] In June 2023, deSouza resigned as CEO of Illumina, and was replaced by interim CEO Charles Dadswell, the company's general counsel.[44] Also in June 2023, Hologic CEO Stephen Macmillan was named non-executive Chairman of the Board of Directors.[45] In September 2023, Agilent Technologies' senior vice president Jacob Thaysen was appointed CEO.[46] In October 2023, the European Commission ordered Grail to be divested from Illumina within the next twelve months.[47] Illumina said it would explore a third-party sale or a capital markets transaction if it fails to win its ongoing challenge in court.[48] |
イ
ルミナ(Illumina,
Inc.)は、アメリカ合衆国のカリフォルニア州サンディエゴに本社を置くバイオテクノロジー企業である。1998年4月1日に法人化されたイルミナは、
遺伝的多様性と生物学的機能の解析のための統合システムの開発、製造、販売を行っている。同社は、シーケンス、遺伝子タイピング、遺伝子発現、プロテオミ
クス市場向けの製品とサービスを提供しており、155カ国以上で事業を展開している。 イルミナの顧客には、ゲノム研究センター、製薬会社、学術機関、臨床研究機関、バイオテクノロジー企業などが含まれる。[1] 沿革 1998年夏、イルミナのオフィスにて、Czarnik、Stuelpnagel、Chee イルミナは、1998年4月にDavid Walt、Larry Bock、John Stuelpnagel、Anthony Czarnik、Mark Cheeによって設立された。ボックとステュエルプネゲルは、ベンチャーキャピタル企業CWグループに勤務していた際に、タフツ大学で後にイルミナの BeadArray技術となるものを発見し[3]、その技術の独占ライセンスの交渉を行った。1999年、イルミナはオリゴヌクレオチドの高処理合成技術 を獲得するために、スパイダー・インスツルメンツ社(ミハエル・レブル、リチャード・ホーテン、ユッタ・アイヒラーにより設立)を買収した。イルミナは 2000年7月に新規株式公開を完了した。[4] イルミナは2001年に一塩基多型(SNP)ジェノタイピングサービスの提供を開始し、2002年にはGoldenGateジェノタイピング技術を使用し た最初のシステムであるイルミナBeadLabを発売した。現在イルミナは、SNPジェノタイピング、遺伝子発現、タンパク質解析など、拡大する遺伝子解 析シーケンスの分野を対象に、マイクロアレイベースの製品およびサービスを提供している。イルミナの技術は、世界中の幅広い学術機関、政府機関、製薬会 社、バイオテクノロジー企業、その他の主要機関で使用されている。 2007年1月26日、同社はイギリスの企業であるソレクサ社を約6億5000万ドルで買収した。ソレクサ社は、1998年6月にシャンカル・バラスブラ マニアムとデビッド・クレナーマンによって、ケンブリッジ大学で発明されたゲノム配列決定技術の開発と商業化を目的として設立された。2005年、 Solexa Ltd.はヘイワードのLynx Therapeuticsに吸収合併され、Solexa, Inc.が設立された。[6]イルミナも、1997年にパスカル・メイヤーとローラン・ファレネリによって発明されたDNAコロニーシークエンシング技術 を使用している。[7]この技術は、2004年にManteia Predictive Medicineからソレクサが取得したものである。この技術は、全ゲノムの再シークエンシング、遺伝子発現解析、および小リボ核酸(sRNA)解析を含 む、さまざまな解析に使用されている。 2009年6月、イルミナは、深度30Xの独自のパーソナル全ゲノムシークエンシングサービスの開始を発表した。[8] 2010年までは、イルミナは「研究用のみ」と表示された機器のみを販売していたが、2010年初頭に、イルミナは臨床検査で使用する BeadXpressシステムについてFDAの承認を取得した。[9][10]これは、当時、同社がCLIAラボを独自に開設し、臨床遺伝子検査を自ら提 供し始めるという戦略の一環であった。[11] イルミナは2011年1月11日、ウィスコンシン州マディソンに拠点を置くEpicentre Biotechnologiesを買収した。[12] 2012年1月25日、ホフマン・ラ・ロシュはイルミナの株式1株あたり44.50ドル、総額約57億ドルでの買収を提案した。約57億ドルであった。 [13][14] ロシュは、買収額を51.00ドル(約68億ドル)に引き上げるなど、他の戦術を試みた。[15] イルミナは買収提案を拒否し、[16] ロシュは4月に買収提案を断念した。[17] 2014年、同社は数百万ドルの製品HiSeq X Tenを発表した。[18][19] 2014年1月時点で、イルミナはすでにゲノムシーケンシングマシンの市場の70%を占めていた。[20] イルミナの機械は、生成されたDNAデータの90%以上を占めていた 。2020年には、同社は非上場企業であるEnancioの買収に投資した。Enancioは、イルミナのデータを特にターゲットとしたDNAデータ圧縮 アルゴリズムを開発しており、ストレージのフットプリントを80%削減できる(例えば、50GBを10GBに圧縮できる)可能性がある。 2016年7月5日、1999年よりCEOを務めていたジェイ・フラットリーが取締役会の執行会長に就任した。2013年より社長を務めていたフランシス・デスーザがCEOを兼任することになった。 2015年後半、イルミナは血流中の癌腫瘍の血液検査に焦点を当てた企 業Grailをスピンオフさせた。2017年、Grailは2回目の資金調達ラウンドで10億ドルの調達を計画し、ビル・ゲイツとジェフ・ベゾスがシリー ズAの資金調達に1億ドルを投資し、イルミナはGrailの20%の株式を維持した Grailの株式を20%保有している。[24] Grailは、ミネソタ州とウィスコンシン州でマンモグラフィー検診を受ける予定の12万人以上の女性を対象とした血液検査試験を実施しているほか、メイ ヨー・クリニックとも提携している。Grailは検査にイルミナのシーケンシング技術を使用している。[25] Grailは2019年までに検査を展開する計画であった。[26] 2020年9月、イルミナはGrailを80億ドルで買収する現金および株式取引案を発表した。[27][28] 2018年11月、イルミナはパシフィック・バイオサイエンシズの買収を1株あたり8.00ドル、総額約12億ドルで提案した。[29][30] 2019年12月、連邦取引委員会(FTC)は買収を阻止するために提訴した。[31] 2020年1月2日、この提案された取引は中止され、イルミナはパシフィックに9800万ドルの解約料を支払った。[32] 2021年3月、FTCはイルミナによるGrailとの71億ドルの垂直統合を阻止するために提訴した。[33][34] 2021年7月、欧州委員会はイルミナによるGrail買収に関する詳細な調査を開始した。[35] 米国のFTCとEU欧州委員会の両方による積極的な調査の命令に反して イルミナは2021年8月18日にグレイルの買収を完了したことを公表した。[36] FTC は直後にイルミナに合併の「解消」を促し[37]、2021年10月には欧州委員会がイルミナにグレイルを別会社として維持するよう命じ[38]、 競争への悪影響を防ぐための暫定措置を採択し、EU合併規則第15条および第14条に基づき、それぞれ平均日次売上高の5%までの制裁金、および/または 年間世界売上高の10%までの罰金を科す可能性があるとした。[35] 2022年9月、米国の行政裁判官は、 独占禁止法上の理由から買収を阻止しようとしたFTCの取り組みを却下した。[39] 2023年4月、FTCはGrailをIlluminaから売却するよう命じた。[40] 2023年7月、欧州委員会はEUの承認なしにGrailの買収を完了したとして、Illuminaに4億3200万ユーロ(4億7600万ドル)の制裁 金を科した。[41] 2022年9月、イルミナはNovaSeq XとNovaSeq X Plusを発表した(→NextSeq 1000 & NextSeq 2000)。 [42] NovaSeq X Plusは、イルミナの以前の機種では年間7,500だったのに対し、年間20,000のゲノム配列決定が可能であり、1回の実行につき最大16テラバイ トのデータを生成できる。[42] このシリーズには、常温で出荷できる再開発された試薬、色素、およびポリメラーゼが含まれている。[43] 2023年6月、デスーザはイルミナのCEOを辞任し、同社の法務顧問であるチャールズ・ダッズウェルが暫定CEOに就任した。[44] 同じく2023年6月、ホロジックのCEOスティーブン・マクミランが取締役会の非常勤会長に就任した。[45] 2023年9月、アジレント・テクノロジーの上級副社長ジェイコブ・セイセンがCEOに任命された。[46] 2023年10月、欧州委員会は、今後12か月以内にイルミナからグレイルを売却するよう命じた。[47] イルミナは、現在係争中の裁判で敗訴した場合、第三者への売却または資本市場取引を検討するとしている。[48] |
| Acquisition history |
買収史(省略) |
| Products DNA sequencing Main articles: Illumina (Solexa) sequencing and Illumina dye sequencing Illumina MiSeq sequencer Illumina sells a number of high-throughput DNA sequencing systems, also known as DNA sequencers, based on technology developed by Solexa. The technology features bridge amplification to generate clusters and reversible terminators for sequence determination.[49][50] The technology behind these sequencing systems involves ligation of fragmented DNA to a chip, followed by primer addition and sequential fluorescent dNTP incorporation and detection. Depending on the kit used, according to the company the MiSeq Series generates up to 25 million reads per run.[51] With dual flow cells, the NextSeq 2000 generates up to 2.4 billion single reads per run[52] and the NovaSeq X Series generates up to 52 billion single reads per run.[53] Illumina uses next-generation sequencing, which is far faster and more efficient than traditional Sanger sequencing.[54] Illumina sequencers perform short-read sequencing, and are image based, utilizing Illumina dye sequencing.[54] This technology has a higher accuracy than long-read sequencing.[54] Flow cells MiSeq Flow Cell (Top) NovaSeq Flow Cell Illumina sequencing happens within the flow cells. These flow cells are small in size and are housed in the flow cell compartment. Flow cell clustering happens when a denatured DNA sample is placed in a flow cell. Primers already in the flow cell channel capture and bind to the ends of the short denatured DNA sample. Then, DNA polymerase is added and the DNA building blocks are introduced. This results in a newly synthesized strand constrained to the bottom of the flow cell. Next, the original template strand is washed out binding the newly synthesized strand to the other DNA sequence present on the surface. DNA polymerase and building blocks are introduced again forming a new strand. These steps are repeated until about 1,000 copies are made in a cluster.[54] |
製品 DNAシークエンシング 主な記事:イルミナ(ソレクサ)シークエンシングおよびイルミナ色素シークエンシング イルミナMiSeqシーケンサー イルミナは、ソレクサが開発した技術を基に、DNAシーケンサーとも呼ばれる多数のハイスループットDNAシーケンシングシステムを販売している。この技 術は、クラスター生成のためのブリッジ増幅と、配列決定のための可逆的ターミネーターを特徴とする。[49][50] これらのシーケンシングシステムを支える技術は、断片化されたDNAをチップに結合させ、続いてプライマーを添加し、蛍光dNTPを逐次的に取り込み、検 出するというものである。 使用するキットによって異なるが、同社によるとMiSeqシリーズは1回の実行につき最大2500万リードを生成する。[51]デュアルフローセルを使用 するNextSeq 2000は1回の実行につき最大24億のシングルリードを生成し[52]、NovaSeq Xシリーズは1回の実行につき最大520億のシングルリードを生成する リードを生成する。[53]イルミナは次世代シーケンシングを使用しており、これは従来のサンガーシーケンシングよりもはるかに高速で効率的である。 [54]イルミナシーケンサーはショートリードシーケンシングを行い、イメージベースで、イルミナ色素シーケンシングを利用している。[54]このテクノ ロジーは、ロングリードシーケンシングよりも精度が高い。[54] フローセル MiSeqフローセル(上) NovaSeqフローセル イルミナシーケンシングはフローセル内で実行される。これらのフローセルは小型で、フローセルコンパートメントに収容されている。変性DNAサンプルがフ ローセルに配置されると、フローセルのクラスタリングが開始される。フローセルチャネルにすでに存在するプライマーが、短い変性DNAサンプルの両端を捕 捉し結合する。次に、DNAポリメラーゼが添加され、DNAビルディングブロックが導入される。その結果、新たに合成された鎖がフローセルの底面に固定さ れる。次に、元のテンプレート鎖が洗い流され、新たに合成された鎖が表面に存在する他のDNA配列に結合する。DNAポリメラーゼとビルディングブロック が再び導入され、新たな鎖が形成される。これらのステップは、約1,000コピーがクラスター内で作成されるまで繰り返される。[54] |
| Litigation Czarnik suit against Illumina In 2005, co-founder and former Chief Scientific Officer Anthony Czarnik sued Illumina. In the case, Czarnik v. Illumina Inc., the trial court granted Illumina's motion to dismiss in part but allowed Czarnik's correction of inventorship claims to continue.[55] Cornell University and Life Technologies suit against Illumina In 2010, Cornell University and Life Technologies filed a lawsuit against Illumina, alleging that its microarray products infringed on eight patents held by the university and exclusively licensed to the start-up. The case was settled in April 2017 without any finding of fault. In September 2017 both parties asked to have the settlement reviewed, with Cornell accusing both Illumina and Life Technologies of misrepresentation and fraud.[56] Cornell claimed that ThermoFisher had promised to settle the suit with Illumina and asked for the Markman wording to be dropped so that it could file a subsequent suit involving other patents invented at Cornell. Instead of filing the suit, ThermoFisher and Illumina settled another lawsuit in California and secretly sublicensed those very same patents. In 2018, Dr. Monib Zirvi filed a lawsuit in the Southern District of New York against Illumina and some of its key employees claiming that they knowingly incorporated ideas and ZipCode DNA sequences invented in the Barany Lab in Illumina's patent applications. Although this suit was dismissed, it was only after Illumina and its attorneys claimed that some of those IP misappropriation were “storm warnings” and thus statutes of limitations had run out on those particular claims. Dr. Monib Zirvi also filed a FOIA case in New Jersey in 2020 for unredacted copies for key NIH grants that Illumina filed early in its existence. William Noon, an in-house attorney at Illumina, had filed a FOIA request for 4 of these key grants as well in January 2015. Patent infringement suits Illumina was a party in a patent lawsuit against competitor Ariosa Diagnostics. The litigation began in 2012 with Verinata Health filing suit against Ariosa. Illumina joined the suit after acquiring Verinata in 2013. Ariosa subsequently brought a counterclaim against Illumina.[57] The trial court granted summary judgment in favor of Ariosa, but the United States Court of Appeals for the Federal Circuit reversed.[58] Ariosa initially pursued an appeal to the Supreme Court of the United States, but the two parties resolved the dispute before the Court decided whether to take the case.[59] In February 2016, Illumina filed a lawsuit against Oxford Nanopore Technologies. Illumina claimed that Oxford Nanopore infringed its patents on the use of a biological nanopore, Mycobacterium smegmatis porinA (MspA), for sequencing systems.[60][61] In August 2016 the parties settled their lawsuit.[62] In February 2020, Illumina filed a patent infringement suit against BGI relating to its "CoolMPS" sequencing products.[63] In return BGI has filed patent infringement lawsuits for violation of federal antitrust and California unfair competition laws, claiming use of "fraudulent behavior" to obtain or enforce sequencing patents that it has asserted against BGI, preventing the firm from entering the US market.[64] However, in May 2022, Illumina was ordered to pay $333 million to a U.S. unit of BGI in California for infringing two patents of DNA-sequencing systems. The jury of the case also said that Illumina willfully infringed the patents, and that their former accusation of BGI's infringement was invalid.[65] On May 6, 2022, a jury in the U.S. District Court for the District of Delaware rendered a verdict that Illumina willfully infringed two patents owned by Complete Genomics, and awarded approximately $334 million to CGI in past damages. The jury also invalidated three patents owned by Illumina.[66] Trade secrets suit against Eltoukhy and Talasaz In March 2022, Illumina sued Helmy Eltoukhy and Amir Talasaz, the co-founders of Guardant, over stealing trade secrets.[67] Guardant called the lawsuit "frivolous and retaliatory" and framed it as a response to its concerns about the Illumina-Grail merger.[67] Guardant also claimed the lawsuit was filed in order to suppress competition in the marketplace.[68] |
訴訟 Czarnik 対イルミナ訴訟 2005年、共同創設者で元最高科学責任者のアンソニー・チャルニックがイルミナを提訴した。Czarnik v. Illumina Inc.事件では、裁判所はイルミナの訴えの一部を退ける申し立てを認めたが、発明者に関するチャルニックの主張は継続することを認めた。 コーネル大学とライフテクノロジーズによるイルミナに対する訴訟 2010年、コーネル大学とライフテクノロジーズはイルミナに対して訴訟を起こし、イルミナのマイクロアレイ製品が同大学が保有し、新興企業に独占的にラ イセンス供与された8件の特許を侵害していると主張した。この訴訟は2017年4月に過失の認定なしに和解した。2017年9月、両当事者は和解の見直し を求めた。コーネル大学はイルミナとライフテクノロジーズの両社に対して虚偽の陳述と詐欺を非難した。[56] コーネル大学は、サーモフィッシャーがイルミナとの訴訟の和解を約束し、コーネル大学で発明された他の特許に関する訴訟を提起できるように、マークマンの 文言を削除するよう求めたと主張した。サーモフィッシャーとイルミナは訴訟を提起する代わりに、カリフォルニア州で別の訴訟を解決し、極秘裏にそれらの まったく同じ特許をサブライセンスした。2018年、モニブ・ジルビ博士は、イルミナおよびその主要な従業員数名を相手取り、イルミナの特許申請に、バ ラーニ研究室で発明されたアイデアおよびZipCode DNA 配列が故意に組み込まれたと主張して、ニューヨーク南部地区で訴訟を起こした。この訴訟は却下されたが、イルミナおよびその弁護士が、これらの知的財産の 不正流用は「嵐の警告」であり、したがってこれらの特定の主張については時効が成立していると主張した後のことだった。また、モニブ・ジルビ博士は 2020年にニュージャージー州で、イルミナが創業初期に申請したNIHの主要助成金の非編集版コピーを要求する情報公開法に基づく訴訟を起こした。イル ミナの社内弁護士であるウィリアム・ヌーンは、2015年1月にこれら主要助成金の4件についても情報公開法に基づく請求を行っていた。 特許侵害訴訟 イルミナは、競合他社であるAriosa Diagnostics社に対する特許訴訟の当事者であった。この訴訟は、Verinata Health社がAriosa社を提訴した2012年に始まった。イルミナは2013年にベリナータを買収した後、訴訟に加わった。その後、アリオサはイ ルミナに対して反訴を起こした。[57] 裁判所はアリオサを支持する略式判決を下したが、連邦巡回区控訴裁判所はこれを覆した。[58] アリオサは当初、米国最高裁判所に上訴したが、裁判所が訴訟の受理を決定する前に両当事者は紛争を解決した。[59] 2016年2月、イルミナはオックスフォード・ナノポア・テクノロジーズを相手取って訴訟を起こした。イルミナは、オックスフォード・ナノポアが生物学的 ナノポアであるMycobacterium smegmatis porinA (MspA) のシーケンシングシステムへの利用に関する特許を侵害していると主張した。[60][61] 2016年8月、両者は訴訟で和解した。[62] 2020年2月、イルミナはBGIのシーケンシング製品「CoolMPS」に関連して特許侵害訴訟を起こした。[63] これに対してBGIは、連邦独占禁止法およびカリフォルニア州不正競争防止法違反で特許侵害訴訟を起こし、 特許を取得または行使し、BGIが米国市場に参入することを妨害したと主張している。[64] しかし、2022年5月、イルミナはDNAシーケンシングシステムの2件の特許を侵害したとして、カリフォルニア州のBGI米国法人に3億3300万ドル の支払いを命じられた。この訴訟の陪審は、イルミナが故意に特許を侵害したとし、BGIの侵害を主張した以前の主張は無効であるとも述べた。[65] 2022年5月6日、デラウェア地区連邦地方裁判所の陪審は、イルミナが故意にコンプリート・ゲノミクスが所有する2件の特許を侵害したとの評決を下し、 CGIに約3億3400万ドルの過去の損害賠償を認めた。陪審はまた、イルミナが所有する3件の特許を無効とした。[66] エルトゥーキーとタラサズに対する営業秘密訴訟 2022年3月、イルミナはガードアントの共同創業者であるヘルミー・エルトウキーとアミール・タラサズを営業秘密の窃盗で訴えた。[67] ガードアントは、この訴訟を「軽率で報復的なもの」と呼び、イルミナとグレイルの合併に対する懸念への対応であると主張した。[67] ガードアントはまた、この訴訟は市場での競争を抑制するために起こされたものであると主張した。[68] |
| https://en.wikipedia.org/wiki/Illumina,_Inc. |
リ ンク
文 献
そ の他の情報
Copyleft, CC, Mitzub'ixi Quq Chi'j, 1996-2099
![]()
☆
 ☆
☆