
第三世代シーケンシング
Third-generation sequencing
☆ 次世代シーケンシング(NGS)、大規模並列シーケンシング、ディープシーケンシングは、ゲノム研究に革命をもたらしたDNAシーケンシング技術を説明す る関連用語である。NGSを使用すれば、ヒトゲノム全体を1日以内に配列決定することができる。これに対し、ヒトゲノム解読に使用された従来のサンガー シークエンシング技術では、最終草案の完成までに10年以上を要した。ゲノム研究では、NGSが従来のサンガーシークエンシングに取って代 わるケースがほ とんどであるが、臨床現場での日常的な使用にはまだ至っていない(→「ゲノミクス」)。
★【関連項目】「サンガー・シーケンシング(第一世代シーケンシング)」→「大規模並列シーケンシング(第二世代シーケンシング)」→「第三世代シーケンシング」→「次世代シーケンシング」
★
以下は、まず、第三世代シークエンシングについて解説する。
| Third-generation
sequencing (also known as long-read sequencing) is a class of DNA
sequencing methods which produce longer sequence reads, under active
development since 2008.[1] Third generation sequencing technologies have the capability to produce substantially longer reads than second generation sequencing, also known as next-generation sequencing.[1] Such an advantage has critical implications for both genome science and the study of biology in general. However, third generation sequencing data have much higher error rates than previous technologies, which can complicate downstream genome assembly and analysis of the resulting data.[2] These technologies are undergoing active development and it is expected that there will be improvements to the high error rates. For applications that are more tolerant to error rates, such as structural variant calling, third generation sequencing has been found to outperform existing methods, even at a low depth of sequencing coverage.[3] Current technologies Sequencing technologies with a different approach than second-generation platforms were first described as "third-generation" in 2008–2009.[4] There are several companies currently at the heart of third generation sequencing technology development, namely, Pacific Biosciences, Oxford Nanopore Technology, Quantapore (CA-USA), and Stratos (WA-USA). These companies are taking fundamentally different approaches to sequencing single DNA molecules. PacBio developed the sequencing platform of single molecule real time sequencing (SMRT), based on the properties of zero-mode waveguides. Signals are in the form of fluorescent light emission from each nucleotide incorporated by a DNA polymerase bound to the bottom of the zL well. Oxford Nanopore’s technology involves passing a DNA molecule through a nanoscale pore structure and then measuring changes in electrical field surrounding the pore; while Quantapore has a different proprietary nanopore approach. Stratos Genomics spaces out the DNA bases with polymeric inserts, "Xpandomers", to circumvent the signal to noise challenge of nanopore ssDNA reading. Also notable is Helicos's single molecule fluorescence approach, but the company entered bankruptcy in the fall of 2015. |
第3世代シーケンシング(ロングリードシーケンシングとも呼ばれる)
は、より長いシーケンスリードを生成するDNAシーケンシング手法の一種であり、2008年より活発に開発が進められている。 第3世代シーケンシング技術は、第2世代シーケンシングよりもはるかに長いリードを生成する能力があり、次世代シーケンシングとも呼ばれる。このような利 点は、ゲノム科学および生物学全般の研究にとって非常に重要な意味を持つ。しかし、第3世代シーケンシングデータは、以前の技術よりもエラー率がはるかに 高く、その結果、下流のゲノムアセンブリやデータ解析が複雑になる可能性がある。[2] これらの技術は現在も活発に開発が進められており、エラー率の改善が期待されている。 構造多型検出など、エラー率に寛容なアプリケーションでは、シーケンスカバレッジの深度が低い場合でも、第3世代シーケンシングが既存の方法よりも優れた 性能を発揮することが分かっている。[3] 現在の技術 第二世代プラットフォームとは異なるアプローチによるシーケンシング技術は、2008年から2009年にかけて初めて「第三世代」として説明された。 [4] 現在、第三世代シーケンシング技術開発の中心となっている企業は、Pacific Biosciences、Oxford Nanopore Technology、Quantapore(カリフォルニア州)、Stratos(ワシントン州)などである。これらの企業は、単一DNA分子のシーケ ンシングに対して、根本的に異なるアプローチを取っている。 PacBioはゼロモード導波路の特性を基に、単分子リアルタイムシーケンシング(SMRT)のシーケンシングプラットフォームを開発した。シグナルは、 zLウェルの底に結合したDNAポリメラーゼによって組み込まれた各ヌクレオチドからの蛍光発光の形をとる。 オックスフォード・ナノポア社の技術は、DNA分子をナノスケールの孔構造に通し、孔の周囲の電界の変化を測定するものである。一方、クオンタペーは独自 のナノポアアプローチを採用している。ストラトス・ジェノミクス社は、ナノポアssDNA読み取りのシグナル対ノイズの課題を回避するために、DNA塩基 をポリマー挿入物「Xpandomers」で間隔を空けて配置している。 また、Helicosの単分子蛍光アプローチも注目に値するが、同社は2015年秋に破産した。 |
| Advantages Longer reads In comparison to the current generation of sequencing technologies, third generation sequencing has the obvious advantage of producing much longer reads. It is expected that these longer read lengths will alleviate numerous computational challenges surrounding genome assembly, transcript reconstruction, and metagenomics among other important areas of modern biology and medicine.[1] It is well known that eukaryotic genomes including primates and humans are complex and have large numbers of long repeated regions. Short reads from second generation sequencing must resort to approximative strategies in order to infer sequences over long ranges for assembly and genetic variant calling. Pair end reads have been leveraged by second generation sequencing to combat these limitations. However, exact fragment lengths of pair ends are often unknown and must also be approximated as well. By making long reads lengths possible, third generation sequencing technologies have clear advantages. Epigenetics Epigenetic markers are stable and potentially heritable modifications to the DNA molecule that are not in its sequence. An example is DNA methylation at CpG sites, which has been found to influence gene expression. Histone modifications are another example. The current generation of sequencing technologies rely on laboratory techniques such as ChIP-sequencing for the detection of epigenetic markers. These techniques involve tagging the DNA strand, breaking and filtering fragments that contain markers, followed by sequencing. Third generation sequencing may enable direct detection of these markers due to their distinctive signal from the other four nucleotide bases.[5] Portability and speed  MinION Portable Gene Sequencer, Oxford Nanopore Technologies Other important advantages of third generation sequencing technologies include portability and sequencing speed.[6] Since minimal sample preprocessing is required in comparison to second generation sequencing, smaller equipments could be designed. Oxford Nanopore Technology has recently commercialized the MinION sequencer. This sequencing machine is roughly the size of a regular USB flash drive and can be used readily by connecting to a laptop. In addition, since the sequencing process is not parallelized across regions of the genome, data could be collected and analyzed in real time. These advantages of third generation sequencing may be well-suited in hospital settings where quick and on-site data collection and analysis is demanded. |
利点 より長いリード 現在のシーケンシング技術と比較すると、第3世代シーケンシング技術には、より長いリードを生成できるという明白な利点がある。これらのより長いリード長 により、ゲノムアセンブリ、転写産物の再構成、メタゲノム解析など、現代の生物学や医学の重要な分野における数多くの計算上の課題が軽減されることが期待 されている。 霊長類やヒトを含む真核生物のゲノムは複雑であり、多数の長い繰り返し領域を持つことはよく知られている。第2世代シーケンサーによる短いリードでは、ア センブリや遺伝子変異の検出のために長距離の配列を推定するには近似的な戦略に頼らざるを得ない。これらの制限に対処するために、第2世代シーケンサーで はペアエンドリードが活用されている。しかし、ペアエンドの断片の長さが正確にわからないことが多く、これも近似的に推定しなければならない。長距離リー ドの長さを可能にする第3世代シーケンサー技術には、明確な利点がある。 エピジェネティクス エピジェネティックマーカーは、DNA分子の配列には存在しない、安定した潜在的に遺伝可能な修飾である。その例としては、遺伝子発現に影響を与えること が分かっているCpG部位におけるDNAメチル化がある。ヒストン修飾もその例である。現在のシーケンシング技術では、エピジェネティックマーカーの検出 にChIP-sequencingなどの実験室技術に依存している。これらの技術では、DNA鎖にタグ付けを行い、マーカーを含む断片を切断およびフィル タリングし、その後シーケンスを行う。第3世代のシーケンシングでは、他の4つのヌクレオチド塩基とは異なるこれらのマーカーのシグナルにより、これらの マーカーを直接検出できる可能性がある。[5] 可搬性とスピード MinIONポータブル遺伝子シーケンサー、オックスフォード・ナノポア・テクノロジー 第3世代シーケンシング技術のその他の重要な利点として、可搬性とシーケンシングのスピードが挙げられる。[6] 第2世代シーケンシングと比較して、サンプルの前処理が最小限で済むため、より小型の機器を設計できる。オックスフォード・ナノポア・テクノロジーは最 近、MinION シーケンサーを商品化した。このシーケンサーは通常のUSBフラッシュドライブとほぼ同じ大きさで、ノートパソコンに接続するだけで使用できる。さらに、 シーケンシングプロセスはゲノムの領域ごとに並列化されていないため、データをリアルタイムで収集し、分析することができる。このような第3世代シーケン サーの利点は、迅速なデータ収集と分析が求められる病院の現場に適しているかもしれない。 |
| Challenges Parts of this article (those related to long-read sequencing technologies producing low-accuracy reads. While true 5 years ago, circular consensus reads with the PacBio Sequel II long-read sequencer can easily achieve an even higher read accuracy than hybrid genome assembly with a combination of other sequencers. [1] PMID 31885515, 28364362, 31406327, 31897449, 31483244) need to be updated. Please help update this article to reflect recent events or newly available information. (January 2020) Third generation sequencing, as of 2008, faced important challenges mainly surrounding accurate identification of nucleotide bases; error rates were still much higher compared to second generation sequencing.[2] This is generally due to instability of the molecular machinery involved. For example, in PacBio’s single molecular and real time sequencing technology, the DNA polymerase molecule becomes increasingly damaged as the sequencing process occurs.[2] Additionally, since the process happens quickly, the signals given off by individual bases may be blurred by signals from neighbouring bases. This poses a new computational challenge for deciphering the signals and consequently inferring the sequence. Methods such as Hidden Markov Models, for example, have been leveraged for this purpose with some success.[5] On average, different individuals of the human population share about 99.9% of their genes. In other words, approximately only one out of every thousand bases would differ between any two person. The high error rates involved with third generation sequencing are inevitably problematic for the purpose of characterizing individual differences that exist between members of the same species.[citation needed] |
課題 この記事の一部(低精度リードを生成する長鎖シーケンス技術に関連する部分。5年前は事実であったが、PacBio Sequel II長鎖シーケンサーによる環状コンセンサスリードは、他のシーケンサーを組み合わせたハイブリッドゲノムアセンブリよりもさらに高いリード精度を簡単に 達成できる。[1] PMID 31885515, 28364362, 31406327, 31897449, 31483244) は更新する必要がある。最近の出来事や新たに利用可能になった情報を反映させるため、この記事を更新するのを手伝ってください。 (2020年1月) 2008年時点での第3世代シーケンシングは、主にヌクレオチド塩基の正確な同定を巡る重要な課題に直面していた。エラー率は第2世代シーケンシングと比 較して依然としてはるかに高かった。[2] これは一般的に、関与する分子機械の不安定性によるものである。例えば、PacBioの単分子リアルタイムシーケンシング技術では、シーケンシングプロセ スが進むにつれ、DNAポリメラーゼ分子が損傷を受ける度合いが高くなる。[2] さらに、プロセスが迅速に進むため、個々の塩基から発せられるシグナルが、近隣の塩基からのシグナルによってぼやけてしまう可能性がある。このため、シグ ナルを解読し、結果として配列を推定するという新たな計算上の課題が生じる。例えば、隠れマルコフモデルなどの手法が、この目的のために活用され、ある程 度の成果を上げている。[5] 平均すると、ヒトの個体は約99.9%の遺伝子を共有している。言い換 えれば、2人のヒトの間では、約1000塩基対につき1つだけが異なることになる。第三世代シーケンサーのエラー率の高さは、同じ種に属する個体間の違い を特徴づけるという目的においては、避けられない問題である。[要出典] |
| Genome assembly Genome assembly is the reconstruction of whole genome DNA sequences. This is generally done with two fundamentally different approaches. Reference alignment When a reference genome is available, as one is in the case of human, newly sequenced reads could simply be aligned to the reference genome in order to characterize its properties. Such reference based assembly is quick and easy but has the disadvantage of “hiding" novel sequences and large copy number variants. In addition, reference genomes do not yet exist for most organisms. De novo assembly De novo assembly is the alternative genome assembly approach to reference alignment. It refers to the reconstruction of whole genome sequences entirely from raw sequence reads. This method would be chosen when there is no reference genome, when the species of the given organism is unknown as in metagenomics, or when there exist genetic variants of interest that may not be detected by reference genome alignment. Given the short reads produced by the current generation of sequencing technologies, de novo assembly is a major computational problem. It is normally approached by an iterative process of finding and connecting sequence reads with sensible overlaps. Various computational and statistical techniques, such as de bruijn graphs and overlap layout consensus graphs, have been leveraged to solve this problem. Nonetheless, due to the highly repetitive nature of eukaryotic genomes, accurate and complete reconstruction of genome sequences in de novo assembly remains challenging. Pair end reads have been posed as a possible solution, though exact fragment lengths are often unknown and must be approximated.[7] 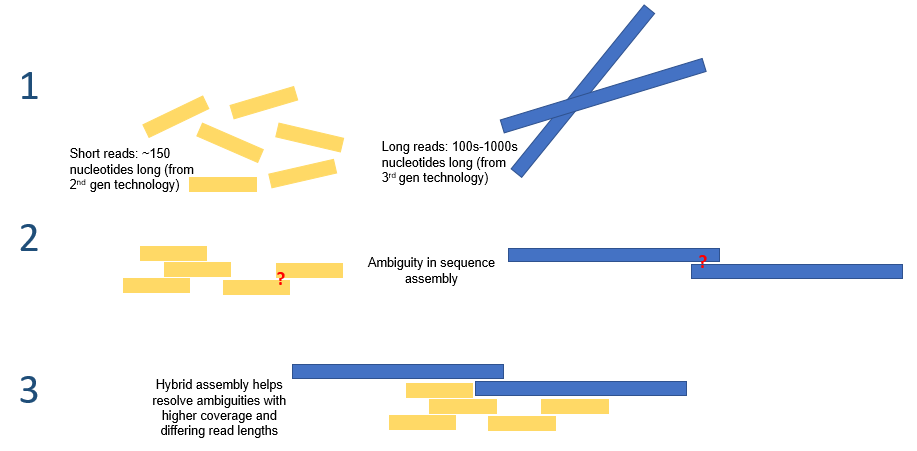 Hybrid assembly – the use of reads from 3rd gen sequencing platforms with shorts reads from 2nd gen platforms – may be used to resolve ambiguities that exist in genomes previously assembled using second generation sequencing. Short second generation reads have also been used to correct errors that exist in the long third generation reads. Hybrid assembly Long read lengths offered by third generation sequencing may alleviate many of the challenges currently faced by de novo genome assemblies. For example, if an entire repetitive region can be sequenced unambiguously in a single read, no computation inference would be required. Computational methods have been proposed to alleviate the issue of high error rates. For example, in one study, it was demonstrated that de novo assembly of a microbial genome using PacBio sequencing alone performed superior to that of second generation sequencing.[8] Third generation sequencing may also be used in conjunction with second generation sequencing. This approach is often referred to as hybrid sequencing. For example, long reads from third generation sequencing may be used to resolve ambiguities that exist in genomes previously assembled using second generation sequencing. On the other hand, short second generation reads have been used to correct errors in that exist in the long third generation reads. In general, this hybrid approach has been shown to improve de novo genome assemblies significantly.[9] |
ゲノムアセンブリ ゲノムアセンブリとは、ゲノムDNA配列全体の再構築を指す。これは一般的に、根本的に異なる2つのアプローチによって行われる。 参照アラインメント 参照ゲノムが利用可能な場合、ヒトの場合のように、新たに配列決定されたリードをその特性を明らかにするために参照ゲノムに単純にアラインメントすること ができる。このような参照ベースのアセンブリは迅速かつ容易であるが、新規配列やコピー数変異の「隠蔽」という欠点がある。さらに、ほとんどの生物には、 参照ゲノムはまだ存在していない。 デノボアセンブリ デノボアセンブリは、参照アラインメントの代替となるゲノムアセンブリのアプローチである。これは、生配列リードからゲノム配列全体を再構築することを指 す。この方法は、参照ゲノムが存在しない場合、メタゲノムのように生物種が不明である場合、または参照ゲノムアラインメントでは検出されない可能性がある 注目すべき遺伝的多型が存在する場合に選択される。 現行のシーケンシング技術で生成される短いリードを考慮すると、デノボアセンブリは大きな計算上の問題となる。通常、これは、意味のあるオーバーラップを 持つシーケンスリードを見つけ、つなぎ合わせる反復プロセスによって対処される。デ・ブルイーングラフやオーバーラップレイアウトコンセンサスグラフなど のさまざまな計算および統計的手法が、この問題の解決に活用されてきた。しかし、真核生物ゲノムの反復性の高さを考慮すると、デノボアセンブリによるゲノ ム配列の正確かつ完全な再構築は依然として困難である。ペアエンドリードは解決策のひとつとして考えられているが、正確な断片の長さは不明であることが多 く、近似値でなければならない。[7] ハイブリッドアセンブリ(第2世代プラットフォームのショートリードと第3世代プラットフォームのリードを併用する)は、第2世代シーケンシングでアセン ブルされたゲノムに存在するあいまいさを解決するために使用される可能性がある。また、第2世代シーケンシングのショートリードは、第3世代シーケンシン グのロングリードに存在するエラーを修正するために使用されることもある。 ハイブリッドアセンブリ 第3世代シーケンシングが提供する長いリード長は、現在デノボゲノムアセンブリが直面している多くの課題を軽減する可能性がある。例えば、反復領域全体が 1回のリードで明確にシーケンスできる場合、計算による推論は必要ない。高いエラー率の問題を軽減するための計算方法が提案されている。例えば、ある研究 では、PacBioシーケンシングのみを使用した微生物ゲノムのデノボアセンブリが、第2世代シーケンシングよりも優れていることが実証された。 第3世代シーケンサーは、第2世代シーケンサーと併用されることもある。このアプローチはハイブリッドシーケンシングと呼ばれることが多い。例えば、第3 世代シーケンサーによる長いリードは、第2世代シーケンサーで以前にアセンブルされたゲノムに存在する曖昧性を解決するために使用される。一方、第2世代 シーケンサーの短いリードは、第3世代シーケンサーの長いリードに存在するエラーを修正するために使用されている。一般的に、このハイブリッドアプローチ は、デノボゲノムアセンブリを大幅に改善することが示されている。[9] |
| Epigenetic markers DNA methylation (DNAm) – the covalent modification of DNA at CpG sites resulting in attached methyl groups – is the best understood component of epigenetic machinery. DNA modifications and resulting gene expression can vary across cell types, temporal development, with genetic ancestry, can change due to environmental stimuli and are heritable. After the discovery of DNAm, researchers have also found its correlation to diseases like cancer and autism.[10] In this disease etiology context DNAm is an important avenue of further research. Advantages The current most common methods for examining methylation state require an assay that fragments DNA before standard second generation sequencing on the Illumina platform. As a result of short read length, information regarding the longer patterns of methylation are lost.[5] Third generation sequencing technologies offer the capability for single molecule real-time sequencing of longer reads, and detection of DNA modification without the aforementioned assay.[11]  PacBio SMRT technology and Oxford Nanopore can use unaltered DNA to detect methylation. Oxford Nanopore Technologies’ MinION has been used to detect DNAm. As each DNA strand passes through a pore, it produces electrical signals which have been found to be sensitive to epigenetic changes in the nucleotides, and a hidden Markov model (HMM) was used to analyze MinION data to detect 5-methylcytosine (5mC) DNA modification.[5] The model was trained using synthetically methylated E. coli DNA and the resulting signals measured by the nanopore technology. Then the trained model was used to detect 5mC in MinION genomic reads from a human cell line which already had a reference methylome. The classifier has 82% accuracy in randomly sampled singleton sites, which increases to 95% when more stringent thresholds are applied.[5] Other methods address different types of DNA modifications using the MinION platform. Stoiber et al. examined 4-methylcytosine (4mC) and 6-methyladenine (6mA), along with 5mC, and also created software to directly visualize the raw MinION data in a human-friendly way.[12] Here they found that in E. coli, which has a known methylome, event windows of 5 base pairs long can be used to divide and statistically analyze the raw MinION electrical signals. A straightforward Mann-Whitney U test can detect modified portions of the E. coli sequence, as well as further split the modifications into 4mC, 6mA or 5mC regions.[12] It seems likely that in the future, MinION raw data will be used to detect many different epigenetic marks in DNA. PacBio sequencing has also been used to detect DNA methylation. In this platform, the pulse width – the width of a fluorescent light pulse – corresponds to a specific base. In 2010 it was shown that the interpulse distance in control and methylated samples are different, and there is a "signature" pulse width for each methylation type.[11] In 2012 using the PacBio platform the binding sites of DNA methyltransferases were characterized.[13] The detection of N6-methylation in C Elegans was shown in 2015.[14] DNA methylation on N6-adenine using the PacBio platform in mouse embryonic stem cells was shown in 2016.[15] Other forms of DNA modifications – from heavy metals, oxidation, or UV damage – are also possible avenues of research using Oxford Nanopore and PacBio third generation sequencing. Drawbacks Processing of the raw data – such as normalization to the median signal – was needed on MinION raw data, reducing real-time capability of the technology.[12] Consistency of the electrical signals is still an issue, making it difficult to accurately call a nucleotide. MinION has low throughput; since multiple overlapping reads are hard to obtain, this further leads to accuracy problems of downstream DNA modification detection. Both the hidden Markov model and statistical methods used with MinION raw data require repeated observations of DNA modifications for detection, meaning that individual modified nucleotides need to be consistently present in multiple copies of the genome, e.g. in multiple cells or plasmids in the sample. For the PacBio platform, too, depending on what methylation you expect to find, coverage needs can vary. As of March 2017, other epigenetic factors like histone modifications have not been discoverable using third-generation technologies. Longer patterns of methylation are often lost because smaller contigs still need to be assembled. |
エピジェネティックマーカー DNAメチル化(DNAm)は、CpG部位における共有結合によるDNAの修飾であり、メチル基の付着を伴う。これは、エピジェネティック機構の最もよく 理解されている要素である。DNAの修飾とそれによる遺伝子発現は、細胞の種類や時間的発達、遺伝的背景によって異なり、環境刺激によって変化し、遺伝性 がある。DNAmの発見後、研究者らは癌や自閉症などの疾患との相関関係も発見している。[10] この疾患の病因という観点では、DNAmはさらなる研究の重要な道筋となる。 利点 現在のメチル化状態を調べる最も一般的な方法では、イルミナプラットフォームの標準的な第2世代シーケンシングの前に、DNAを断片化するアッセイが必要 である。短いリード長により、より長いメチル化パターンに関する情報が失われる。第3世代シーケンシング技術は、前述のアッセイを必要とせずに、より長い リードの単分子リアルタイムシーケンシングとDNA修飾の検出を行うことができる。 PacBio SMRT技術とOxford Nanoporeは、修飾されていないDNAを用いてメチル化を検出することができる。 Oxford Nanopore TechnologiesのMinIONは、DNAmの検出に使用されている。DNA鎖が孔を通過する際に電気信号が発生し、その信号はヌクレオチドのエ ピジェネティックな変化に敏感であることが分かっている。隠れマルコフモデル(HMM)を使用してMinIONデータを分析し、5-メチルシトシン (5mC)DNA修飾を検出した。[5] このモデルは、合成的にメチル化された大腸菌DNAとナノポア技術で測定した結果の信号を使用して訓練された。その後、この学習済みモデルを使用して、参 照メチルームがすでに存在するヒト細胞株の MinION ゲノムリードから 5mC を検出した。この分類器は、ランダムに抽出された単一サイトにおいて 82% の精度を示し、より厳格な閾値を適用すると 95% にまで向上した。[5] MinION プラットフォームを使用して、他の手法では異なるタイプの DNA 修飾に対応している。Stoiber 氏らは、5mC とともに 4-メチルシトシン(4mC)と 6-メチルアデニン(6mA)を調べ、MinION の生データを直接、人間にとってわかりやすい形で視覚化するソフトウェアも作成した。[12] ここでは、メチル化状態が既知である大腸菌において、5 塩基対のイベントウィンドウを使用して MinION の生電気信号を分割し、統計的に分析できることがわかった。単純なマン・ホイットニーのU検定によって、大腸菌の配列の修飾部分を検出できるだけでなく、 さらに修飾部分を4mC、6mA、または5mC領域に分割することもできる。[12] 今後、MinIONの生データが、DNAのさまざまなエピジェネティックなマーカーを検出するために使用される可能性が高い。 PacBioシーケンシングもまた、DNAメチル化の検出に使用されている。このプラットフォームでは、蛍光パルスの幅であるパルス幅が特定の塩基に対応 する。2010年には、コントロールサンプルとメチル化サンプルにおけるパルス間の距離が異なり、各メチル化タイプに「シグネチャー」パルス幅があること が示された。[11] 2012年には、PacBioプラットフォームを使用して、DNAメチルトランスフェラーゼの結合部位が 特性が明らかにされた。[13] 2015年には、CエレガンスにおけるN6-メチル化の検出が示された。[14] 2016年には、マウス胚性幹細胞におけるPacBioプラットフォームを用いたN6-アデニン上のDNAメチル化が示された。[15] 重金属、酸化、または紫外線による損傷など、他のDNA修飾の形態も、Oxford NanoporeとPacBioの第3世代シーケンサーを使用した研究の対象となり得る。 欠点 MinIONの生データでは、中央値信号への正規化などの生データの処理が必要であり、技術のリアルタイム能力が低下していた。[12] 電気信号の一貫性は依然として問題であり、ヌクレオチドを正確に特定することが難しい。MinIONはスループットが低く、複数のオーバーラップするリー ドを取得することが困難であるため、下流のDNA修飾検出の精度に問題が生じる。MinIONの生データで使用される隠れマルコフモデルと統計的手法は、 検出のためにDNA修飾の繰り返し観察を必要とするため、個々の修飾ヌクレオチドは、サンプル内の複数の細胞またはプラスミドなど、ゲノムの複数のコピー に一貫して存在する必要がある。 PacBioプラットフォームの場合も、予想されるメチル化の種類によって、カバレッジのニーズは異なる。2017年3月現在、ヒストン修飾のような他の エピジェネティック因子は、第3世代の技術では検出できない。より長いメチル化パターンは、より小さなコンティグをアセンブルする必要があるため、しばし ば失われる。 |
| Transcriptomics Transcriptomics is the study of the transcriptome, usually by characterizing the relative abundances of messenger RNA molecules in the tissue under study. According to the central dogma of molecular biology, genetic information flows from double stranded DNA molecules to single stranded mRNA molecules where they can be readily translated into functional protein molecules. By studying the transcriptome, one can gain valuable insight into the regulation of gene expression. While expression levels can be more or less accurately depicted by second generation sequencing (we can assume that actual abundances of the population of transcripts are randomly sampled), transcript-level information still remains an important challenge.[16] As a consequence, the role of alternative splicing in molecular biology remains largely elusive. Third generation sequencing technologies hold promising prospects in resolving this issue by enabling sequencing of mRNA molecules at their full lengths. Alternative splicing Alternative splicing (AS) is the process by which a single gene may give rise to multiple distinct mRNA transcripts and consequently different protein translations.[17] Some evidence suggests that AS is a ubiquitous phenomenon and may play a key role in determining the phenotypes of organisms, especially in complex eukaryotes; all eukaryotes contain genes consisting of introns that may undergo AS. In particular, it has been estimated that AS occurs in 95% of all human multi-exon genes.[18] AS has undeniable potential to influence myriad biological processes. Advancing knowledge in this area has critical implications for the study of biology in general. Transcript reconstruction The current generation of sequencing technologies produce only short reads, putting tremendous limitation on the ability to detect distinct transcripts; short reads must be reverse engineered into original transcripts that could have given rise to the resulting read observations.[19] This task is further complicated by the highly variable expression levels across transcripts, and consequently variable read coverages across the sequence of the gene.[19] In addition, exons may be shared among individual transcripts, rendering unambiguous inferences essentially impossible.[17] Existing computational methods make inferences based on the accumulation of short reads at various sequence locations often by making simplifying assumptions.[19] Cufflinks takes a parsimonious approach, seeking to explain all the reads with the fewest possible number of transcripts.[20] On the other hand, StringTie attempts to simultaneously estimate transcript abundances while assembling the reads.[19] These methods, while reasonable, may not always identify real transcripts. A study published in 2008 surveyed 25 different existing transcript reconstruction protocols.[16] Its evidence suggested that existing methods are generally weak in assembling transcripts, though the ability to detect individual exons are relatively intact.[16] According to the estimates, average sensitivity to detect exons across the 25 protocols is 80% for Caenorhabditis elegans genes.[16] In comparison, transcript identification sensitivity decreases to 65%. For human, the study reported an exon detection sensitivity averaging to 69% and transcript detection sensitivity had an average of a mere 33%.[16] In other words, for human, existing methods are able to identify less than half of all existing transcript. Third generation sequencing technologies have demonstrated promising prospects in solving the problem of transcript detection as well as mRNA abundance estimation at the level of transcripts. While error rates remain high, third generation sequencing technologies have the capability to produce much longer read lengths.[21] Pacific Bioscience has introduced the iso-seq platform, proposing to sequence mRNA molecules at their full lengths.[21] It is anticipated that Oxford Nanopore will put forth similar technologies. The trouble with higher error rates may be alleviated by supplementary high quality short reads. This approach has been previously tested and reported to reduce the error rate by more than 3 folds.[22] |
トランスクリプトミクス トランスクリプトミクスは、通常、研究対象の組織におけるメッセンジャーRNA分子の相対的豊富度を特徴付けることによって、トランスクリプトームを研究 するものである。分子生物学の中心ドグマによると、遺伝情報は二重らせんDNA分子から単一鎖mRNA分子へと流れ、そこで容易に機能性タンパク質分子へ と翻訳される。トランスクリプトームを研究することによって、遺伝子発現の制御に関する貴重な洞察を得ることができる。 第二世代シーケンシングでは、発現レベルをある程度正確に描写することができる(転写産物の集団の実際の存在量がランダムにサンプリングされていると仮定 できる)が、転写レベルの情報は依然として重要な課題である。[16] その結果、分子生物学における選択的スプライシングの役割は依然としてほとんど解明されていない。第三世代シーケンシング技術は、mRNA分子の全長シー ケンシングを可能にすることで、この問題の解決に有望な見通しをもたらしている。 選択的スプライシング 選択的スプライシング(AS)とは、1つの遺伝子が複数の異なるmRNA転写物を生み出し、その結果、異なるタンパク質翻訳が起こるプロセスである。 [17] いくつかの証拠から、ASは普遍的な現象であり、特に複雑な真核生物において生物の表現型を決定する上で重要な役割を果たしている可能性があることが示唆 されている。すべての真核生物は、ASを受ける可能性があるイントロンから構成される遺伝子を含んでいる。特に、ヒトの多エクソン遺伝子の95%でASが 起こっていると推定されている。[18] ASは、数多くの生物学的プロセスに影響を及ぼす可能性を否定できない。この分野の知識の進歩は、生物学全般の研究に重大な影響を及ぼす。 転写産物の再構成 現行のシーケンシング技術では短いリードしか生成されないため、個々の転写産物を検出する能力には大きな限界がある。短いリードは、結果として得られた リードの観察結果を生み出した可能性のある元の転写産物にリバースエンジニアリングしなければならない。[19] この作業は、転写産物間の発現レベルが大きく異なるため、さらに複雑になる。その結果、遺伝子の配列全体にわたってリードのカバレッジが異なる。[19] さらに、エクソンは個々の転写産物間で共有されている可能性があり、明確な推論は基本的に不可能である。[ 17] 既存の計算方法は、さまざまな配列位置における短いリードの蓄積に基づいて推論を行うが、その際、単純化された仮定を置くことが多い。[19] Cufflinksは、最小限の転写産物で全てのリードを説明しようとする、倹約的なアプローチを取る。[20] 一方、StringTieは、リードをアセンブルしながら、転写産物の存在量を同時に推定しようとする。[19] これらの方法は妥当ではあるが、常に実際の転写産物を特定できるとは限らない。 2008年に発表された研究では、25種類の異なる既存の転写産物再構成プロトコルが調査された。[16] その証拠によると、既存の方法は転写産物のアセンブリには概して弱いものの、個々のエクソンの検出能力は比較的損なわれていないことが示唆された。 [16] 推定によると、25種類のプロトコル全体におけるエクソン検出の平均感度は、線虫の遺伝子では80%であった。[16] これに対し、転写産物同定の感度は65%に減少する。ヒトの場合、この研究ではエクソン検出感度が平均69%、トランスクリプト検出感度はわずか33%と いう結果であった。[16] 言い換えれば、ヒトの場合、既存の方法では全トランスクリプトの半分以下しか同定できないということである。 第三世代シーケンシング技術は、転写産物の検出および転写産物のレベルでのmRNAの存在量の推定という問題の解決に有望な見通しを示している。エラー率 は依然として高いものの、第三世代シーケンシング技術は、はるかに長いリード長を生成する能力がある。[21] パシフィック・バイオサイエンス社は、iso-seqプラットフォームを導入し、mRNA分子を全長にわたってシーケンスすることを提案している。 [21] オックスフォード・ナノポア社も同様の技術を開発する見込みである。エラー率の高さという問題は、高品質の短いリードを補足することで緩和できる可能性が ある。このアプローチは以前にもテストされ、エラー率を3分の1以下に削減できることが報告されている。[22] |
| Metagenomics Metagenomics is the analysis of genetic material recovered directly from environmental samples. Advantages The main advantage for third-generation sequencing technologies in metagenomics is their speed of sequencing in comparison to second generation techniques. Speed of sequencing is important for example in the clinical setting (i.e. pathogen identification), to allow for efficient diagnosis and timely clinical actions. Oxford Nanopore's MinION was used in 2015 for real-time metagenomic detection of pathogens in complex, high-background clinical samples. The first Ebola virus (EBOV) read was sequenced 44 seconds after data acquisition.[23] There was uniform mapping of reads to genome; at least one read mapped to >88% of the genome. The relatively long reads allowed for sequencing of a near-complete viral genome to high accuracy (97–99% identity) directly from a primary clinical sample.[23] A common phylogenetic marker for microbial community diversity studies is the 16S ribosomal RNA gene. Both MinION and PacBio's SMRT platform have been used to sequence this gene.[24][25] In this context the PacBio error rate was comparable to that of shorter reads from 454 and Illumina's MiSeq sequencing platforms.[citation needed] Drawbacks MinION's high error rate (~10-40%) prevented identification of antimicrobial resistance markers, for which single nucleotide resolution is necessary. For the same reason, eukaryotic pathogens were not identified.[23] Ease of carryover contamination when re-using the same flow cell (standard wash protocols don’t work) is also a concern. Unique barcodes may allow for more multiplexing. Furthermore, performing accurate species identification for bacteria, fungi and parasites is very difficult, as they share a larger portion of the genome, and some only differ by <5%. The per base sequencing cost is still significantly more than that of MiSeq. However, the prospect of supplementing reference databases with full-length sequences from organisms below the limit of detection from the Sanger approach;[24] this could possibly greatly help the identification of organisms in metagenomics. |
メタゲノミクス メタゲノミクスとは、環境サンプルから直接回収した遺伝物質の解析である。 利点 メタゲノムにおける第3世代シーケンシング技術の主な利点は、第2世代技術と比較したシーケンスのスピードである。シーケンスのスピードは、例えば臨床現 場(すなわち病原体の特定)において、効率的な診断と迅速な臨床対応を可能にするために重要である。 オックスフォード・ナノポアのMinIONは、2015年に複雑でバックグラウンドが高い臨床サンプル中の病原体のリアルタイムメタゲノム検出に使用され た。最初のエボラウイルス(EBOV)のリードは、データ取得から44秒後に配列決定された。[23] リードはゲノムに均一にマッピングされ、少なくとも1つのリードはゲノムの88%以上にマッピングされた。比較的長いリードにより、一次臨床サンプルから 直接、高い精度(同一性97~99%)でほぼ完全なウイルスゲノムのシーケンスが可能となった。 微生物群の多様性研究における一般的な系統発生マーカーは、16SリボソームRNA遺伝子である。MinIONとPacBioのSMRTプラットフォーム の両方が、この遺伝子の配列決定に使用されている。[24][25]この文脈において、PacBioのエラー率は、454およびIlluminaの MiSeqシーケンシングプラットフォームからの短いリードのエラー率と同等であった。[要出典] 欠点 MinIONのエラー率が高い(~10~40%)ため、一塩基分解能が必要な薬剤耐性マーカーの特定が妨げられた。同じ理由で、真核病原体の特定もできな かった。[23] 同じフローセルを再使用する際のキャリーオーバー汚染のしやすさ(標準的な洗浄プロトコルでは対応できない)も懸念事項である。固有のバーコードにより、 多重化の可能性が高まる可能性がある。さらに、細菌、真菌、寄生虫の正確な種同定を行うのは非常に困難である。これらの生物はゲノムの大部分を共有してお り、一部はわずか5%しか異ならないからだ。 塩基配列決定コストは、MiSeqよりも依然としてかなり高い。しかし、サンガー法では検出限界以下の生物の全長配列をリファレンスデータベースに追加す るという見通しがある。[24] これは、メタゲノムにおける生物の同定に大いに役立つ可能性がある。 |
| First-generation sequencing Second-generation sequencing Third-generation_sequencing.html |
第一世代シーケンシング 第二世代シーケンシング 第三世代シーケンシング |
リ ンク
文 献
そ の他の情報
Copyleft, CC, Mitzub'ixi Quq Chi'j, 1996-2099
![]()
☆
 ☆
☆