ゲノミクス
Genomics

ゲノミクス
Genomics
★ゲノミクスは、ゲノムの構造、機能、進化、マッピング、編集に焦点を当てた分子生物学の学
際的分野である。ゲノムとは、生物のDNAの完全な集合であり、すべての遺伝子とその階層的、3次元的な構造構成を含む。個々の遺伝子と遺伝におけるその
役割の研究を指す遺伝学とは対照的に、ゲノミクスは、生物のすべての遺伝子、それらの相互関係、および生物への影響の集合的な特徴づけと定量化を目指す。
その結果、タンパク質は臓器や組織などの身体構造を構成するとともに、化学反応を制御し、細胞間のシグナルを伝達する。ゲノミクスはまた、ゲノム全体の機
能と構造を組み立て、解析するための高スループットDNA配列決定とバイオインフォマティクスの利用によるゲノムの配列決定と解析も含む。ゲノミクスの進
歩は、脳のような最も複雑な生物学的システムの理解を促進するために、発見に基づく研究とシステム生物学における革命を引き起こした。
この分野には、エピスタシス(ある遺伝子が別の遺伝子に及ぼす影響)、プレイオトロピー(1つの遺伝子が複数の形質に影響を及ぼす)、ヘテロシス(雑種強
勢)などのゲノム内(ゲノム内)の現象や、ゲノム内の遺伝子座や対立遺伝子間の相互作用の研究も含まれる。
| Genomics is an interdisciplinary
field of molecular biology focusing on the structure, function,
evolution, mapping, and editing of genomes. A genome is an organism's
complete set of DNA, including all of its genes as well as its
hierarchical, three-dimensional structural configuration.[1][2][3][4]
In contrast to genetics, which refers to the study of individual genes
and their roles in inheritance, genomics aims at the collective
characterization and quantification of all of an organism's genes,
their interrelations and influence on the organism.[5] Genes may direct
the production of proteins with the assistance of enzymes and messenger
molecules. In turn, proteins make up body structures such as organs and
tissues as well as control chemical reactions and carry signals between
cells. Genomics also involves the sequencing and analysis of genomes
through uses of high throughput DNA sequencing and bioinformatics to
assemble and analyze the function and structure of entire
genomes.[6][7] Advances in genomics have triggered a revolution in
discovery-based research and systems biology to facilitate
understanding of even the most complex biological systems such as the
brain.[8] The field also includes studies of intragenomic (within the genome) phenomena such as epistasis (effect of one gene on another), pleiotropy (one gene affecting more than one trait), heterosis (hybrid vigour), and other interactions between loci and alleles within the genome.[9] |
ゲノミクスは、ゲノムの構造、機能、進化、マッピング、編集に焦点を当
てた分子生物学の学際的分野である。ゲノムとは、生物のDNAの完全な集合であり、すべての遺伝子とその階層的、3次元的な構造構成を含む[1][2]
[3][4]。個々の遺伝子と遺伝におけるその役割の研究を指す遺伝学とは対照的に、ゲノミクスは、生物のすべての遺伝子、それらの相互関係、および生物
への影響の集合的な特徴づけと定量化を目指す[5]。その結果、タンパク質は臓器や組織などの身体構造を構成するとともに、化学反応を制御し、細胞間のシ
グナルを伝達する。ゲノミクスはまた、ゲノム全体の機能と構造を組み立て、解析するための高スループットDNA配列決定とバイオインフォマティクスの利用
によるゲノムの配列決定と解析も含む[6][7]。ゲノミクスの進歩は、脳のような最も複雑な生物学的システムの理解を促進するために、発見に基づく研究
とシステム生物学における革命を引き起こした[8]。 この分野には、エピスタシス(ある遺伝子が別の遺伝子に及ぼす影響)、プレイオトロピー(1つの遺伝子が複数の形質に影響を及ぼす)、ヘテロシス(雑種強 勢)などのゲノム内(ゲノム内)の現象や、ゲノム内の遺伝子座や対立遺伝子間の相互作用の研究も含まれる[9]。 |
| History Etymology From the Greek ΓΕΝ[10] gen, "gene" (gamma, epsilon, nu, epsilon) meaning "become, create, creation, birth", and subsequent variants: genealogy, genesis, genetics, genic, genomere, genotype, genus etc. While the word genome (from the German Genom, attributed to Hans Winkler) was in use in English as early as 1926,[11] the term genomics was coined by Tom Roderick, a geneticist at the Jackson Laboratory (Bar Harbor, Maine), over beers with Jim Womack, Tom Shows and Stephen O’Brien at a meeting held in Maryland on the mapping of the human genome in 1986.[12] First as the name for a new journal and then as a whole new science discipline.[13] |
歴史 語源 ギリシャ語のΓΕΝ[10]gen、「なる、創る、創造、誕生」を意味する「遺伝子」(γ、ε、ν、ε)、およびそれに続く変化形:系図、創世記、遺伝 学、genic、genomere、genotype、genusなどから。ゲノムという言葉(ドイツ語のGenomに由来し、ハンス・ウィンクラーによ るとされる)は、早くも1926年には英語で使われていたが[11]、ゲノミクスという言葉は、1986年にメリーランド州で開催されたヒトゲノムのマッ ピングに関する会議で、ジャクソン研究所(メイン州バーハーバー)の遺伝学者であるトム・ロデリックが、ジム・ウォマック、トム・ショウズ、スティーブ ン・オブライエンとビールを飲みながら考えた造語である[12]。 |
|
Early sequencing efforts Following Rosalind Franklin's confirmation of the helical structure of DNA, James D. Watson and Francis Crick's publication of the structure of DNA in 1953 and Fred Sanger's publication of the Amino acid sequence of insulin in 1955, nucleic acid sequencing became a major target of early molecular biologists.[14] In 1964, Robert W. Holley and colleagues published the first nucleic acid sequence ever determined, the ribonucleotide sequence of alanine transfer RNA.[15][16] Extending this work, Marshall Nirenberg and Philip Leder revealed the triplet nature of the genetic code and were able to determine the sequences of 54 out of 64 codons in their experiments.[17] In 1972, Walter Fiers and his team at the Laboratory of Molecular Biology of the University of Ghent (Ghent, Belgium) were the first to determine the sequence of a gene: the gene for Bacteriophage MS2 coat protein.[18] Fiers' group expanded on their MS2 coat protein work, determining the complete nucleotide-sequence of bacteriophage MS2-RNA (whose genome encodes just four genes in 3569 base pairs [bp]) and Simian virus 40 in 1976 and 1978, respectively.[19][20] DNA-sequencing technology developed  Frederick Sanger  Walter Gilbert Frederick Sanger and Walter Gilbert shared half of the 1980 Nobel Prize in Chemistry for Independently developing methods for the sequencing of DNA. In addition to his seminal work on the amino acid sequence of insulin, Frederick Sanger and his colleagues played a key role in the development of DNA sequencing techniques that enabled the establishment of comprehensive genome sequencing projects.[9] In 1975, he and Alan Coulson published a sequencing procedure using DNA polymerase with radiolabelled nucleotides that he called the Plus and Minus technique.[21][22] This involved two closely related methods that generated short oligonucleotides with defined 3' termini. These could be fractionated by electrophoresis on a polyacrylamide gel (called polyacrylamide gel electrophoresis) and visualised using autoradiography. The procedure could sequence up to 80 nucleotides in one go and was a big improvement, but was still very laborious. Nevertheless, in 1977 his group was able to sequence most of the 5,386 nucleotides of the single-stranded bacteriophage φX174, completing the first fully sequenced DNA-based genome.[23] The refinement of the Plus and Minus method resulted in the chain-termination, or Sanger method (see below), which formed the basis of the techniques of DNA sequencing, genome mapping, data storage, and bioinformatic analysis most widely used in the following quarter-century of research.[24][25] In the same year Walter Gilbert and Allan Maxam of Harvard University independently developed the Maxam-Gilbert method (also known as the chemical method) of DNA sequencing, involving the preferential cleavage of DNA at known bases, a less efficient method.[26][27] For their groundbreaking work in the sequencing of nucleic acids, Gilbert and Sanger shared half the 1980 Nobel Prize in chemistry with Paul Berg (recombinant DNA). |
初期の配列決定の取り組み ロザリンド・フランクリンがDNAのらせん構造を確認し、ジェームズ・D・ワトソンとフランシス・クリックが1953年にDNAの構造を発表し、フレッ ド・サンガーが1955年にインスリンのアミノ酸配列を発表したのに続き、核酸配列決定は初期の分子生物学者の主要な標的となった。この研究を発展させ、 マーシャル・ニレンバーグとフィリップ・レダーは、遺伝暗号の三重項性を明らかにし、彼らの実験で64コドンのうち54コドンの配列を決定することができ た[17]。 [1972年、ゲント大学分子生物学研究所(ベルギー、ゲント)のウォルター・フィアーズと彼のチームは、バクテリオファージMS2コートタンパク質の遺 伝子という遺伝子の塩基配列を初めて決定した[18]。フィアーズのグループはMS2コートタンパク質の研究を発展させ、1976年と1978年にそれぞ れバクテリオファージMS2-RNA(そのゲノムは3569塩基対[bp]に4つの遺伝子しかコードしていない)とシミアンウイルス40の完全な塩基配列 を決定した[19][20]。 DNA配列決定技術が開発される フレデリック・サンガー ウォルター・ギルバート フレデリック・サンガーとウォルター・ギルバートは、DNAの塩基配列決定法を独自に開発したとして、1980年のノーベル化学賞の半分を分け合った。 1975年、サンガーとアラン・コールソンは、DNAポリメラーゼと放射性標識ヌクレオチドを用いた塩基配列決定法を発表し、これをプラス・マイナス法と 呼んだ[21][22]。これらはポリアクリルアミドゲル上の電気泳動(ポリアクリルアミドゲル電気泳動と呼ばれる)によって分画され、オートラジオグラ フィーを用いて可視化された。この方法では一度に80塩基までの塩基配列を決定することができ、大きな進歩であったが、それでも非常に手間のかかるもので あった。それでも1977年、彼のグループは一本鎖のバクテリオファージφX174の5,386ヌクレオチドのほとんどを配列決定することができ、初めて DNAベースのゲノムを完全に配列決定した。プラス・マイナス法を改良した結果、連鎖終結法、すなわちサンガー法(下記参照)が生まれ、その後の四半世紀 にわたる研究で最も広く使われたDNA配列決定、ゲノムマッピング、データ保存、バイオインフォマティクス解析の技術の基礎となった。 [24][25]同年、ハーバード大学のウォルター・ギルバートとアラン・マクサムは、既知の塩基におけるDNAの優先的切断を伴う、DNA配列決定のマ クサム・ギルバート法(化学的方法としても知られる)を独自に開発したが、これはあまり効率的な方法ではなかった[26][27]。 核酸の配列決定における画期的な業績により、ギルバートとサンガーは、ポール・バーグ(組み換えDNA)と共に1980年のノーベル化学賞の半分を受賞し た。 |
|
Complete genomes The advent of these technologies resulted in a rapid intensification in the scope and speed of completion of genome sequencing projects. The first complete genome sequence of a eukaryotic organelle, the human mitochondrion (16,568 bp, about 16.6 kb [kilobase]), was reported in 1981,[28] and the first chloroplast genomes followed in 1986.[29][30] In 1992, the first eukaryotic chromosome, chromosome III of brewer's yeast Saccharomyces cerevisiae (315 kb) was sequenced.[31] The first free-living organism to be sequenced was that of Haemophilus influenzae (1.8 Mb [megabase]) in 1995.[32] The following year a consortium of researchers from laboratories across North America, Europe, and Japan announced the completion of the first complete genome sequence of a eukaryote, S. cerevisiae (12.1 Mb), and since then genomes have continued being sequenced at an exponentially growing pace.[33] As of October 2011, the complete sequences are available for: 2,719 viruses, 1,115 archaea and bacteria, and 36 eukaryotes, of which about half are fungi.[34][35]  "Hockey stick" graph showing the exponential growth of public sequence databases. The number of genome projects has increased as technological improvements continue to lower the cost of sequencing. (A) Exponential growth of genome sequence databases since 1995. (B) The cost in US Dollars (USD) to sequence one million bases. (C) The cost in USD to sequence a 3,000 Mb (human-sized) genome on a log-transformed scale. Most of the microorganisms whose genomes have been completely sequenced are problematic pathogens, such as Haemophilus influenzae, which has resulted in a pronounced bias in their phylogenetic distribution compared to the breadth of microbial diversity.[36][37] Of the other sequenced species, most were chosen because they were well-studied model organisms or promised to become good models. Yeast (Saccharomyces cerevisiae) has long been an important model organism for the eukaryotic cell, while the fruit fly Drosophila melanogaster has been a very important tool (notably in early pre-molecular genetics). The worm Caenorhabditis elegans is an often used simple model for multicellular organisms. The zebrafish Brachydanio rerio is used for many developmental studies on the molecular level, and the plant Arabidopsis thaliana is a model organism for flowering plants. The Japanese pufferfish (Takifugu rubripes) and the spotted green pufferfish (Tetraodon nigroviridis) are interesting because of their small and compact genomes, which contain very little noncoding DNA compared to most species.[38][39] The mammals dog (Canis familiaris),[40] brown rat (Rattus norvegicus), mouse (Mus musculus), and chimpanzee (Pan troglodytes) are all important model animals in medical research.[27] A rough draft of the human genome was completed by the Human Genome Project in early 2001, creating much fanfare.[41] This project, completed in 2003, sequenced the entire genome for one specific person, and by 2007 this sequence was declared "finished" (less than one error in 20,000 bases and all chromosomes assembled).[41] In the years since then, the genomes of many other individuals have been sequenced, partly under the auspices of the 1000 Genomes Project, which announced the sequencing of 1,092 genomes in October 2012.[42] Completion of this project was made possible by the development of dramatically more efficient sequencing technologies and required the commitment of significant bioinformatics resources from a large international collaboration.[43] The continued analysis of human genomic data has profound political and social repercussions for human societies.[44] |
完全なゲノム このような技術の出現により、ゲノム配列決定プロジェクトの範囲と完了スピードは急速に強化された。真核生物の小器官であるヒトのミトコンドリア (16,568 bp、約16.6 kb [キロベース])の最初の完全なゲノム配列が1981年に報告され[28]、最初の葉緑体ゲノムが1986年に続いた。 [1992年には、最初の真核生物の染色体、ビール酵母サッカロマイセス・セレビシエのIII番染色体(315kb)の塩基配列が決定された[31]。 翌年、北米、ヨーロッパ、日本の研究者からなるコンソーシアムが、真核生物であるセレビシエ(12.1Mb)の完全なゲノム配列の完成を発表した: 2011年10月現在、2,719のウイルス、1,115の古細菌とバクテリア、36の真核生物(そのうちの約半数は真菌である)についての完全な配列が 利用可能である[34][35]。 公開配列データベースの指数関数的な成長を示す「ホッケースティック」グラフ。 ゲノムプロジェクトの数は、技術的な改善によって配列決定のコストが下がり続けるにつれて増加している。(A)1995年以降のゲノム配列データベースの 指数関数的な増加。(B)100万塩基の塩基配列を決定するのにかかった費用(米ドル)。(C)対数変換した3,000 Mb(ヒトサイズ)のゲノムの配列決定にかかる費用(USD)。 ゲノムが完全に解読された微生物のほとんどは、インフルエンザ菌のような問題のある病原体であり、その結果、微生物の多様性の広さと比較して、系統学的分 布に顕著な偏りが生じている[36][37]。酵母(Saccharomyces cerevisiae)は長い間、真核細胞の重要なモデル生物であり、ミバエ(Drosophila melanogaster)は(特に初期の分子遺伝学において)非常に重要なツールであった。線虫は多細胞生物の単純なモデルとしてよく使われる。ゼブラ フィッシュ(Brachydanio rerio)は分子レベルでの多くの発生研究に使われ、シロイヌナズナ(Arabidopsis thaliana)は顕花植物のモデル生物である。ニホンフグ(Takifugu rubripes)とマダラフグ(Tetraodon nigroviridis)は、ゲノムが小さくコンパクトで、ほとんどの種に比べてノンコーディングDNAをほとんど含まないという点で興味深い[38] [39]。 哺乳類のイヌ(Canis familiaris)、[40]ヒメネズミ(Rattus norvegicus)、マウス(Mus musculus)、チンパンジー(Pan troglodytes)はすべて、医学研究の重要なモデル動物である[27]。 2003年に完了したこのプロジェクトでは、特定の一人のゲノムの全塩基配列が決定され、2007年までにこの塩基配列が「完成した」(20,000塩基 中の1塩基未満の誤りしかなく、すべての染色体が組み合わされている)と宣言された[41]。それ以来数年間で、他の多くの個人のゲノムの塩基配列が決定 され、その一部は1000人ゲノムプロジェクトの支援の下で行われ、2012年10月に1,092のゲノムの塩基配列決定が発表された[42]。 [42]。このプロジェクトの完了は、飛躍的に効率的なシーケンス技術の開発によって可能となり、大規模な国際共同研究による多大なバイオインフォマティ クスリソースの投入を必要とした[43]。ヒトゲノムデータの継続的な解析は、人類社会にとって政治的・社会的に重大な影響を及ぼす[44]。 |
|
The "omics" revolution General schema showing the relationships of the genome, transcriptome, proteome, and metabolome (lipidome) Main articles: Omics and Human proteome project The English-language neologism omics informally refers to a field of study in biology ending in -omics, such as genomics, proteomics or metabolomics. The related suffix -ome is used to address the objects of study of such fields, such as the genome, proteome, or metabolome (lipidome) respectively. The suffix -ome as used in molecular biology refers to a totality of some sort; similarly omics has come to refer generally to the study of large, comprehensive biological data sets. While the growth in the use of the term has led some scientists (Jonathan Eisen, among others[45]) to claim that it has been oversold,[46] it reflects the change in orientation towards the quantitative analysis of complete or near-complete assortment of all the constituents of a system.[47] In the study of symbioses, for example, researchers which were once limited to the study of a single gene product can now simultaneously compare the total complement of several types of biological molecules.[48][49] |
「オミックス」革命 ゲノム、トランスクリプトーム、プロテオーム、メタボローム(リピドーム)の関係を示す一般的な図式。 主な記事 オミックスとヒトプロテオームプロジェクト 英語のomicsという新語は、非公式には、ゲノミクス、プロテオミクス、メタボロミクスなど、-omicsで終わる生物学の研究分野を指す。関連する接 尾辞-omeは、それぞれゲノム、プロテオーム、メタボローム(リピドーム)など、このような分野の研究対象を指すのに使われる。分子生物学で用いられる 接尾辞-omeは、ある種の全体性を意味する。同様に、オミクスは大規模で包括的な生物学的データセットの研究全般を指すようになった。この用語の使用の 増加により、一部の科学者(ジョナサン・アイゼンなど[45])は、この用語は過剰に販売されていると主張するようになったが[46]、システムのすべて の構成要素の完全な、またはほぼ完全な品揃えの定量的分析への方向性の変化を反映している[47]。 |
| Genome analysis Main article: Genome project After an organism has been selected, genome projects involve three components: the sequencing of DNA, the assembly of that sequence to create a representation of the original chromosome, and the annotation and analysis of that representation.[9] 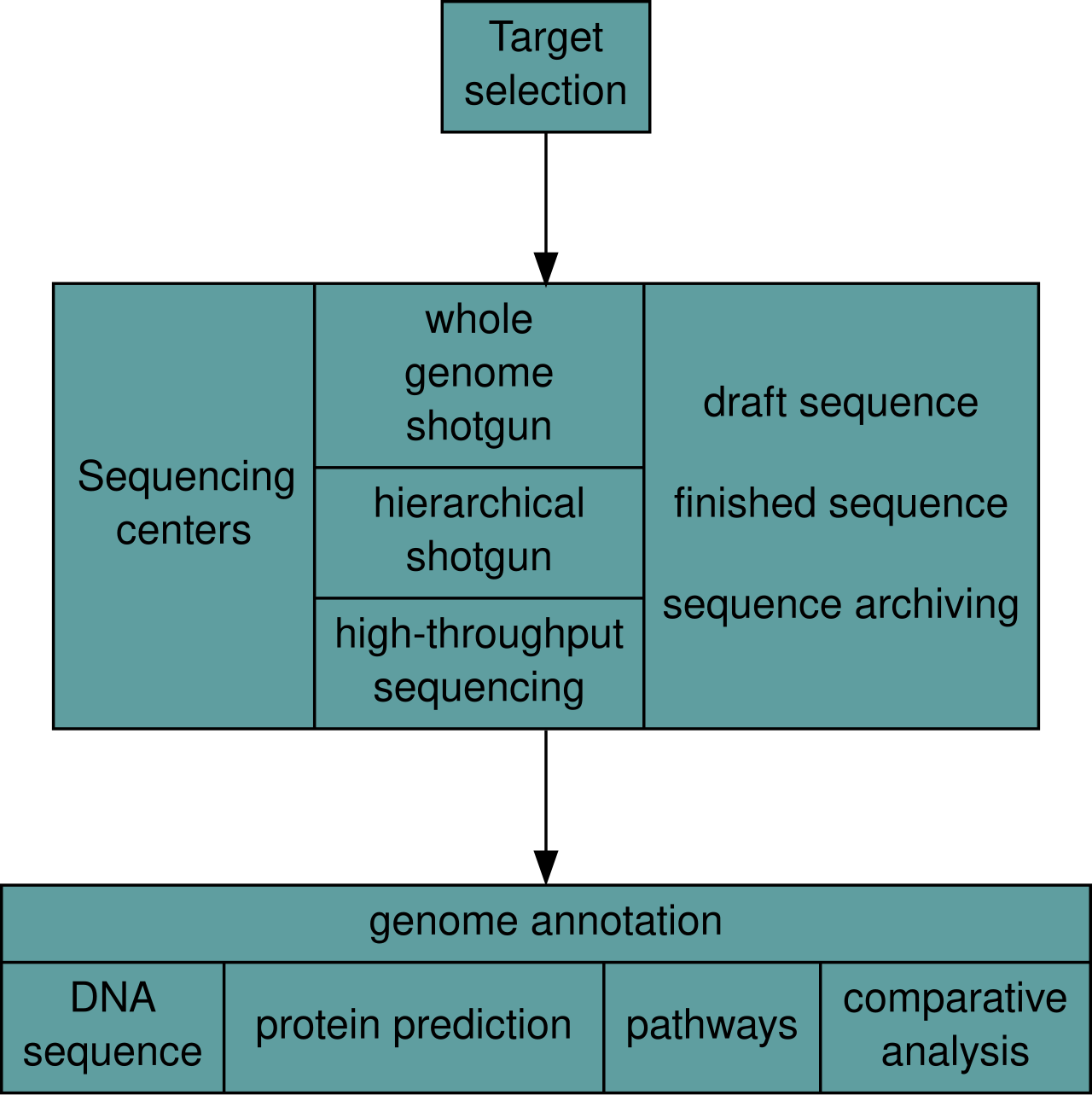 Overview of a genome project. First, the genome must be selected, which involves several factors including cost and relevance. Second, the sequence is generated and assembled at a given sequencing center (such as BGI or DOE JGI). Third, the genome sequence is annotated at several levels: DNA, protein, gene pathways, or comparatively. |
ゲノム解析 主な記事 ゲノムプロジェクト ゲノムプロジェクトには、DNAの塩基配列の決定、その塩基配列を組み立てて元の染色体の表現を作成すること、そしてその表現の注釈付けと解析という3つ の要素が含まれる[9]。 ゲノムプロジェクトの概要 まず、ゲノムを選択しなければならないが、これにはコストや関連性などいくつかの要素が関係する。第二に、所定のシーケンスセンター(BGIやDOE JGIなど)で配列が作成され、組み立てられる。第三に、ゲノム配列はいくつかのレベルでアノテーションされる: DNA、タンパク質、遺伝子パスウェイ、比較などである。 |
| Sequencing Main article: DNA Sequencing Historically, sequencing was done in sequencing centers, centralized facilities (ranging from large independent institutions such as Joint Genome Institute which sequence dozens of terabases a year, to local molecular biology core facilities) which contain research laboratories with the costly instrumentation and technical support necessary. As sequencing technology continues to improve, however, a new generation of effective fast turnaround benchtop sequencers has come within reach of the average academic laboratory.[50][51] On the whole, genome sequencing approaches fall into two broad categories, shotgun and high-throughput (or next-generation) sequencing.[9] Shotgun sequencing  An ABI PRISM 3100 Genetic Analyzer. Such capillary sequencers automated early large-scale genome sequencing efforts. Main article: Shotgun sequencing Shotgun sequencing is a sequencing method designed for analysis of DNA sequences longer than 1000 base pairs, up to and including entire chromosomes.[52] It is named by analogy with the rapidly expanding, quasi-random firing pattern of a shotgun. Since gel electrophoresis sequencing can only be used for fairly short sequences (100 to 1000 base pairs), longer DNA sequences must be broken into random small segments which are then sequenced to obtain reads. Multiple overlapping reads for the target DNA are obtained by performing several rounds of this fragmentation and sequencing. Computer programs then use the overlapping ends of different reads to assemble them into a continuous sequence.[52][53] Shotgun sequencing is a random sampling process, requiring over-sampling to ensure a given nucleotide is represented in the reconstructed sequence; the average number of reads by which a genome is over-sampled is referred to as coverage.[54] For much of its history, the technology underlying shotgun sequencing was the classical chain-termination method or 'Sanger method', which is based on the selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication.[23][55] Recently, shotgun sequencing has been supplanted by high-throughput sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use, primarily for smaller-scale projects and for obtaining especially long contiguous DNA sequence reads (>500 nucleotides).[56] Chain-termination methods require a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleosidetriphosphates (dNTPs), and modified nucleotides (dideoxyNTPs) that terminate DNA strand elongation. These chain-terminating nucleotides lack a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in DNA sequencers.[9] Typically, these machines can sequence up to 96 DNA samples in a single batch (run) in up to 48 runs a day.[57] High-throughput sequencing See also: Illumina dye sequencing and Ion semiconductor sequencing The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once.[58][59] High-throughput sequencing is intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods. In ultra-high-throughput sequencing, as many as 500,000 sequencing-by-synthesis operations may be run in parallel.[60][61]  Illumina Genome Analyzer II System. Illumina technologies have set the standard for high-throughput massively parallel sequencing.[50] The Illumina dye sequencing method is based on reversible dye-terminators and was developed in 1996 at the Geneva Biomedical Research Institute, by Pascal Mayer and Laurent Farinelli.[62] In this method, DNA molecules and primers are first attached on a slide and amplified with polymerase so that local clonal colonies, initially coined "DNA colonies", are formed. To determine the sequence, four types of reversible terminator bases (RT-bases) are added and non-incorporated nucleotides are washed away. Unlike pyrosequencing, the DNA chains are extended one nucleotide at a time and image acquisition can be performed at a delayed moment, allowing for very large arrays of DNA colonies to be captured by sequential images taken from a single camera. Decoupling the enzymatic reaction and the image capture allows for optimal throughput and theoretically unlimited sequencing capacity; with an optimal configuration, the ultimate throughput of the instrument depends only on the A/D conversion rate of the camera. The camera takes images of the fluorescently labeled nucleotides, then the dye along with the terminal 3' blocker is chemically removed from the DNA, allowing the next cycle.[63] An alternative approach, ion semiconductor sequencing, is based on standard DNA replication chemistry. This technology measures the release of a hydrogen ion each time a base is incorporated. A microwell containing template DNA is flooded with a single nucleotide, if the nucleotide is complementary to the template strand it will be incorporated and a hydrogen ion will be released. This release triggers an ISFET ion sensor. If a homopolymer is present in the template sequence multiple nucleotides will be incorporated in a single flood cycle, and the detected electrical signal will be proportionally higher.[64] |
シーケンス 主な記事 DNAシーケンス 歴史的には、シーケンシングはシーケンシングセンターで行われてきた。シーケンシングセンターは、集中型の施設(年間数十テラベースのシーケンシングを行 うJoint Genome Instituteのような大規模な独立機関から、地域の分子生物学コア施設まで)で、高価な装置と必要な技術サポートを備えた研究室を有している。しか し、シーケンシング技術が向上し続けるにつれて、新世代の効果的な高速ターンアラウンドのベンチトップシーケンサーは、平均的な学術研究室にも手が届くよ うになった[50][51]。全体として、ゲノムシーケンシングのアプローチは、ショットガンシーケンシングとハイスループットシーケンシング(または次 世代シーケンシング)の2つに大別される[9]。 ショットガンシーケンス ABI PRISM 3100 Genetic Analyzer。このようなキャピラリーシーケンサーは、初期の大規模ゲノムシーケンス作業を自動化した。 主な記事 ショットガンシーケンス ショットガンシーケンスは、染色体全体を含む1000塩基対より長いDNA配列の解析用に設計されたシーケンス手法である[52]。ショットガンの急速に 拡大する準ランダム発射パターンになぞらえて命名された。ゲル電気泳動シーケンシングはかなり短い配列(100~1000塩基対)にしか使用できないた め、長いDNA配列はランダムな小さなセグメントに分割しなければならない。この断片化と塩基配列決定を数回繰り返すことで、標的DNAについて複数の重 複したリードが得られる。ショットガンシーケンスはランダムサンプリングプロセスであり、所定のヌクレオチドが再構築された配列に確実に含まれるようにす るにはオーバーサンプリングが必要である。 その歴史の大部分において、ショットガンシーケンスの基礎となる技術は古典的な鎖終結法または「サンガー法」であり、これはin vitroでのDNA複製中にDNAポリメラーゼが鎖終結ジデオキシヌクレオチドを選択的に組み込むことに基づく[23][55]。しかし、サンガー法 は、主に小規模なプロジェクトや、特に長い連続DNA配列リード(>500ヌクレオチド)を得るために広く使われている[56]。鎖終結法は、一本 鎖DNAテンプレート、DNAプライマー、DNAポリメラーゼ、通常のデオキシヌクレオシド三リン酸(dNTPs)、およびDNA鎖の伸長を終結させる修 飾ヌクレオチド(ジデオキシNTPs)を必要とする。これらの鎖終結ヌクレオチドは、2つのヌクレオチド間のホスホジエステル結合形成に必要な3'-OH 基を持たないため、ddNTPが組み込まれるとDNAポリメラーゼはDNAの伸長を停止する。ddNTPは、DNAシーケンサーで検出するために、放射性 標識または蛍光標識することができる[9]。通常、これらの装置は、1バッチ(ラン)で最大96のDNAサンプルを、1日に最大48回シーケンスすること ができる[57]。 ハイスループットシーケンス 以下も参照のこと: イルミナ色素シーケンスおよびイオン半導体シーケンス 低コストのシーケンシングに対する高い需要が、シーケンシングプロセスを並列化し、一度に数千から数百万のシーケンスを生成するハイスループットシーケン シング技術の開発を後押ししている[58][59]。ハイスループットシーケンシングは、標準的な色素ターミネーター法で可能な以上にDNAシーケンシン グのコストを下げることを目的としている。超高スループットシーケンスでは、50万ものシーケンスバイシンセシス操作が並行して実行される可能性がある [60][61]。 イルミナGenome Analyzer IIシステム。イルミナの技術は、ハイスループット大量並列シーケンスの標準となっている[50]。 イルミナの色素シーケンス法は、可逆的な色素ターミネーターに基づくもので、1996年にジュネーブ生物医学研究所でPascal MayerとLaurent Farinelliによって開発された[62]。この方法では、まずDNA分子とプライマーをスライド上に付着させ、ポリメラーゼで増幅することで、当初 「DNAコロニー」と呼ばれた局所的なクローンコロニーが形成される。塩基配列を決定するために、4種類の可逆的ターミネーター塩基(RT塩基)が付加さ れ、取り込まれなかったヌクレオチドは洗浄される。パイロシークエンシングとは異なり、DNA鎖は一度に1ヌクレオチドずつ伸長され、画像取得は遅れた瞬 間に行うことができるため、非常に大きなDNAコロニーのアレイを、1台のカメラから撮影した連続画像で捉えることができる。酵素反応と画像キャプチャを 切り離すことで、最適なスループットと理論上無制限のシーケンス能力が可能になる。最適な構成では、装置の究極のスループットはカメラのA/D変換レート にのみ依存する。カメラは蛍光標識されたヌクレオチドの画像を撮影し、その後、末端の3'ブロッカーとともに色素がDNAから化学的に除去され、次のサイ クルが可能になる[63]。 イオン半導体シーケンシングという別のアプローチは、標準的なDNA複製化学に基づいている。この技術は、塩基が組み込まれるたびに水素イオンが放出され ることを測定する。鋳型DNAを含むマイクロウェルに1つのヌクレオチドを流し、そのヌクレオチドが鋳型鎖と相補的であれば取り込まれ、水素イオンが放出 される。この放出がISFETイオンセンサーのトリガーとなる。鋳型配列にホモポリマーが存在する場合、1回のフラッドサイクルで複数のヌクレオチドが取 り込まれ、それに比例して検出される電気信号も高くなる[64]。 |
| Assembly Main article: Sequence assembly 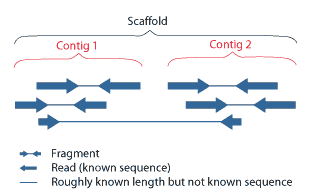 Overlapping reads form contigs; contigs and gaps of known length form scaffolds. 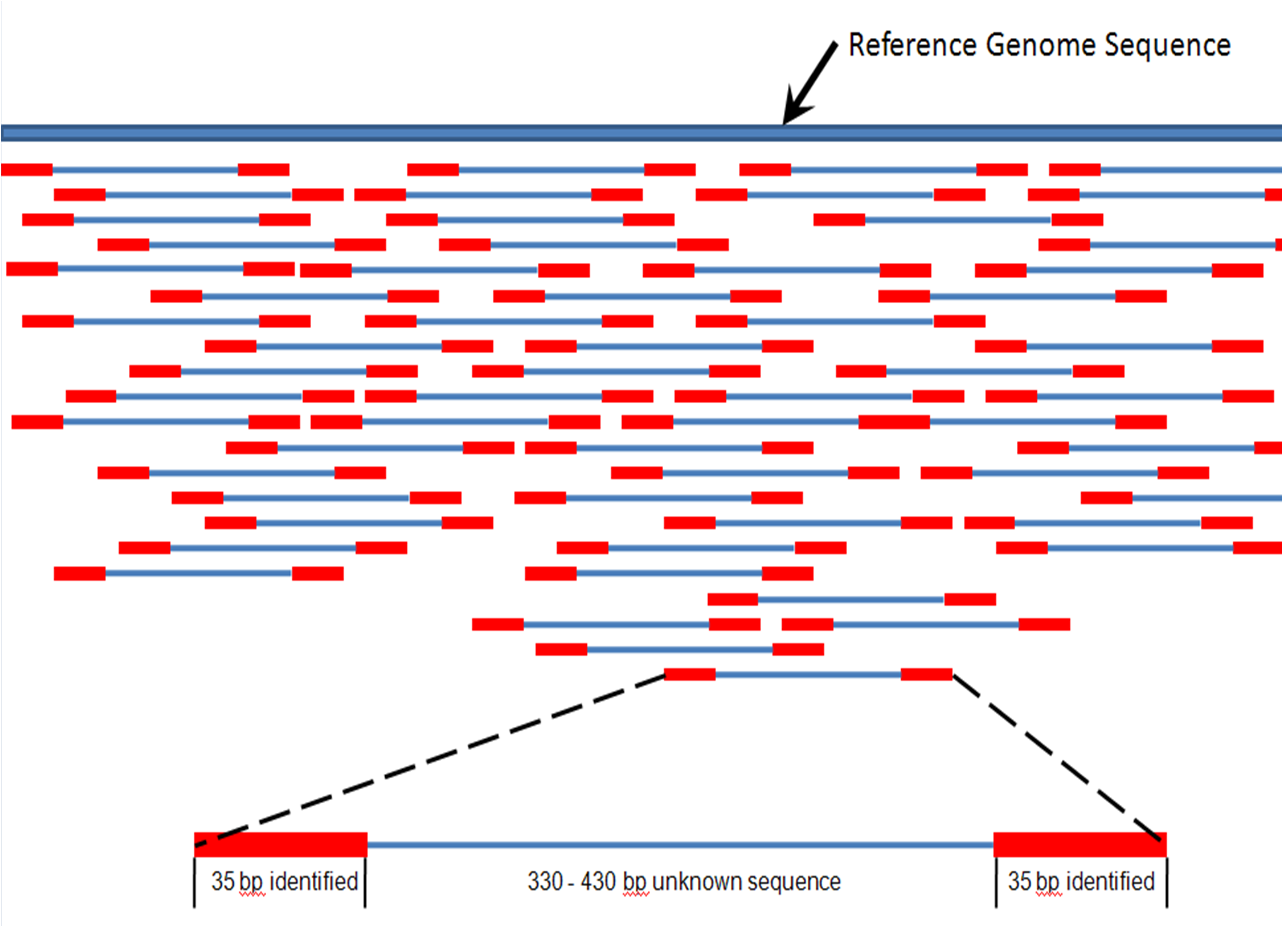 Paired end reads of next generation sequencing data mapped to a reference genome. Multiple, fragmented sequence reads must be assembled together on the basis of their overlapping areas. Sequence assembly refers to aligning and merging fragments of a much longer DNA sequence in order to reconstruct the original sequence.[9] This is needed as current DNA sequencing technology cannot read whole genomes as a continuous sequence, but rather reads small pieces of between 20 and 1000 bases, depending on the technology used. Third generation sequencing technologies such as PacBio or Oxford Nanopore routinely generate sequencing reads 10-100 kb in length; however, they have a high error rate at approximately 1 percent.[65][66] Typically the short fragments, called reads, result from shotgun sequencing genomic DNA, or gene transcripts (ESTs).[9] Assembly approaches Assembly can be broadly categorized into two approaches: de novo assembly, for genomes which are not similar to any sequenced in the past, and comparative assembly, which uses the existing sequence of a closely related organism as a reference during assembly.[54] Relative to comparative assembly, de novo assembly is computationally difficult (NP-hard), making it less favourable for short-read NGS technologies. Within the de novo assembly paradigm there are two primary strategies for assembly, Eulerian path strategies, and overlap-layout-consensus (OLC) strategies. OLC strategies ultimately try to create a Hamiltonian path through an overlap graph which is an NP-hard problem. Eulerian path strategies are computationally more tractable because they try to find a Eulerian path through a deBruijn graph.[54] Finishing Finished genomes are defined as having a single contiguous sequence with no ambiguities representing each replicon.[67] Annotation Main article: Genome annotation The DNA sequence assembly alone is of little value without additional analysis.[9] Genome annotation is the process of attaching biological information to sequences, and consists of three main steps:[68] identifying portions of the genome that do not code for proteins identifying elements on the genome, a process called gene prediction, and attaching biological information to these elements. Automatic annotation tools try to perform these steps in silico, as opposed to manual annotation (a.k.a. curation) which involves human expertise and potential experimental verification.[69] Ideally, these approaches co-exist and complement each other in the same annotation pipeline (also see below). Traditionally, the basic level of annotation is using BLAST for finding similarities, and then annotating genomes based on homologues.[9] More recently, additional information is added to the annotation platform. The additional information allows manual annotators to deconvolute discrepancies between genes that are given the same annotation. Some databases use genome context information, similarity scores, experimental data, and integrations of other resources to provide genome annotations through their Subsystems approach. Other databases (e.g. Ensembl) rely on both curated data sources as well as a range of software tools in their automated genome annotation pipeline.[70] Structural annotation consists of the identification of genomic elements, primarily ORFs and their localisation, or gene structure. Functional annotation consists of attaching biological information to genomic elements. Sequencing pipelines and databases The need for reproducibility and efficient management of the large amount of data associated with genome projects mean that computational pipelines have important applications in genomics.[71] |
アセンブリ 主な記事 配列アセンブリー オーバーラップしたリードはコンティグを形成し、コンティグと既知の長さのギャップはスキャフォールドを形成する。 次世代シーケンスデータのペアエンドリードを参照ゲノムにマッピングする。 断片化された複数のシーケンスリードは、重複する領域を基にアセンブルする必要がある。 配列アセンブリーとは、元の配列を再構築するために、はるかに長いDNA配列の断片を整列させて結合することを指す[9]。現在のDNAシーケンス技術で は、ゲノム全体を連続した配列として読み取ることができず、使用する技術によって20塩基から1000塩基の小さな断片を読み取るため、この作業が必要と なる。PacBioやOxford Nanoporeなどの第3世代シーケンシング技術では、長さ10~100kbのシーケンシングリードを日常的に生成しているが、エラー率は約1%と高い [65][66]。一般的に、リードと呼ばれる短い断片は、ゲノムDNAや遺伝子転写産物(EST)をショットガンでシーケンシングした結果である [9]。 アセンブルアプローチ アセンブルは、過去に配列決定されたゲノムと類似していないゲノムを対象とするde novoアセンブルと、アセンブル時に近縁の生物の既存の配列を参照として使用する比較アセンブルの2つのアプローチに大別される[54]。比較アセンブ ルと比較して、de novoアセンブルは計算が難しく(NP-hard)、ショートリードNGS技術には不向きである。de novoアセンブリーのパラダイムには、オイラーパス戦略とオーバーラップ・レイアウト・コンセンサス(OLC)戦略という2つの主要なアセンブリー戦略 がある。OLC戦略は、最終的にオーバーラップグラフを通るハミルトニアン経路を作成しようとするもので、NP困難問題である。オイラーパス戦略は、 deBruijnグラフを通るオイラーパスを見つけようとするため、計算上より扱いやすい[54]。 フィニッシング 完成したゲノムは、各レプリコンを表すあいまいさのない単一の連続した配列を持つものと定義される[67]。 注釈付け 主な記事 ゲノムアノテーション ゲノムアノテーションとは、配列に生物学的情報を付加するプロセスであり、主に以下の3つのステップからなる[68]。 タンパク質をコードしていないゲノム部分を特定する。 ゲノム上のエレメントを同定する(遺伝子予測と呼ばれるプロセス)。 これらの要素に生物学的情報を付加する。 自動アノテーションツールは、これらのステップをインシリコで実行しようとするものであり、人間の専門知識と潜在的な実験的検証を伴う手動アノテーション (別名キュレーション)とは対照的である[69]。理想的には、これらのアプローチは同じアノテーションパイプラインの中で共存し、互いに補完し合う(下 記も参照)。 伝統的に、基本的なアノテーションレベルは、類似性を見つけるためにBLASTを使用し、相同性に基づいてゲノムをアノテーションすることである。この追 加情報により、同じアノテーションが付与された遺伝子間の矛盾を手作業で解決することができる。データベースによっては、ゲノムコンテキスト情報、類似度 スコア、実験データ、他のリソースの統合を利用して、Subsystemsアプローチでゲノムアノテーションを提供している。他のデータベース (Ensemblなど)は、自動化されたゲノムアノテーションパイプラインにおいて、様々なソフトウェアツールだけでなく、キュレートされたデータソース の両方に依存している[70]。構造アノテーションは、ゲノム要素、主にORFとその局在、または遺伝子構造の同定からなる。機能アノテーションは、ゲノ ム要素に生物学的情報を付加することである。 シーケンスパイプラインとデータベース ゲノムプロジェクトに関連する大量のデータの再現性と効率的な管理の必要性から、計算パイプラインはゲノミクスにおいて重要な用途を持つ[71]。 |
| Research areas Functional genomics Main article: Functional genomics Functional genomics is a field of molecular biology that attempts to make use of the vast wealth of data produced by genomic projects (such as genome sequencing projects) to describe gene (and protein) functions and interactions. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. Functional genomics attempts to answer questions about the function of DNA at the levels of genes, RNA transcripts, and protein products. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "gene-by-gene" approach. A major branch of genomics is still concerned with sequencing the genomes of various organisms, but the knowledge of full genomes has created the possibility for the field of functional genomics, mainly concerned with patterns of gene expression during various conditions. The most important tools here are microarrays and bioinformatics. Structural genomics Main article: Structural genomics  An example of a protein structure determined by the Midwest Center for Structural Genomics Structural genomics seeks to describe the 3-dimensional structure of every protein encoded by a given genome.[72][73] This genome-based approach allows for a high-throughput method of structure determination by a combination of experimental and modeling approaches. The principal difference between structural genomics and traditional structural prediction is that structural genomics attempts to determine the structure of every protein encoded by the genome, rather than focusing on one particular protein. With full-genome sequences available, structure prediction can be done more quickly through a combination of experimental and modeling approaches, especially because the availability of large numbers of sequenced genomes and previously solved protein structures allow scientists to model protein structure on the structures of previously solved homologs. Structural genomics involves taking a large number of approaches to structure determination, including experimental methods using genomic sequences or modeling-based approaches based on sequence or structural homology to a protein of known structure or based on chemical and physical principles for a protein with no homology to any known structure. As opposed to traditional structural biology, the determination of a protein structure through a structural genomics effort often (but not always) comes before anything is known regarding the protein function. This raises new challenges in structural bioinformatics, i.e. determining protein function from its 3D structure.[74] Epigenomics Main article: Epigenomics Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome.[75] Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence (Russell 2010 p. 475). Two of the most characterized epigenetic modifications are DNA methylation and histone modification.[76] Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development[77] and tumorigenesis.[75] The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.[78] Metagenomics 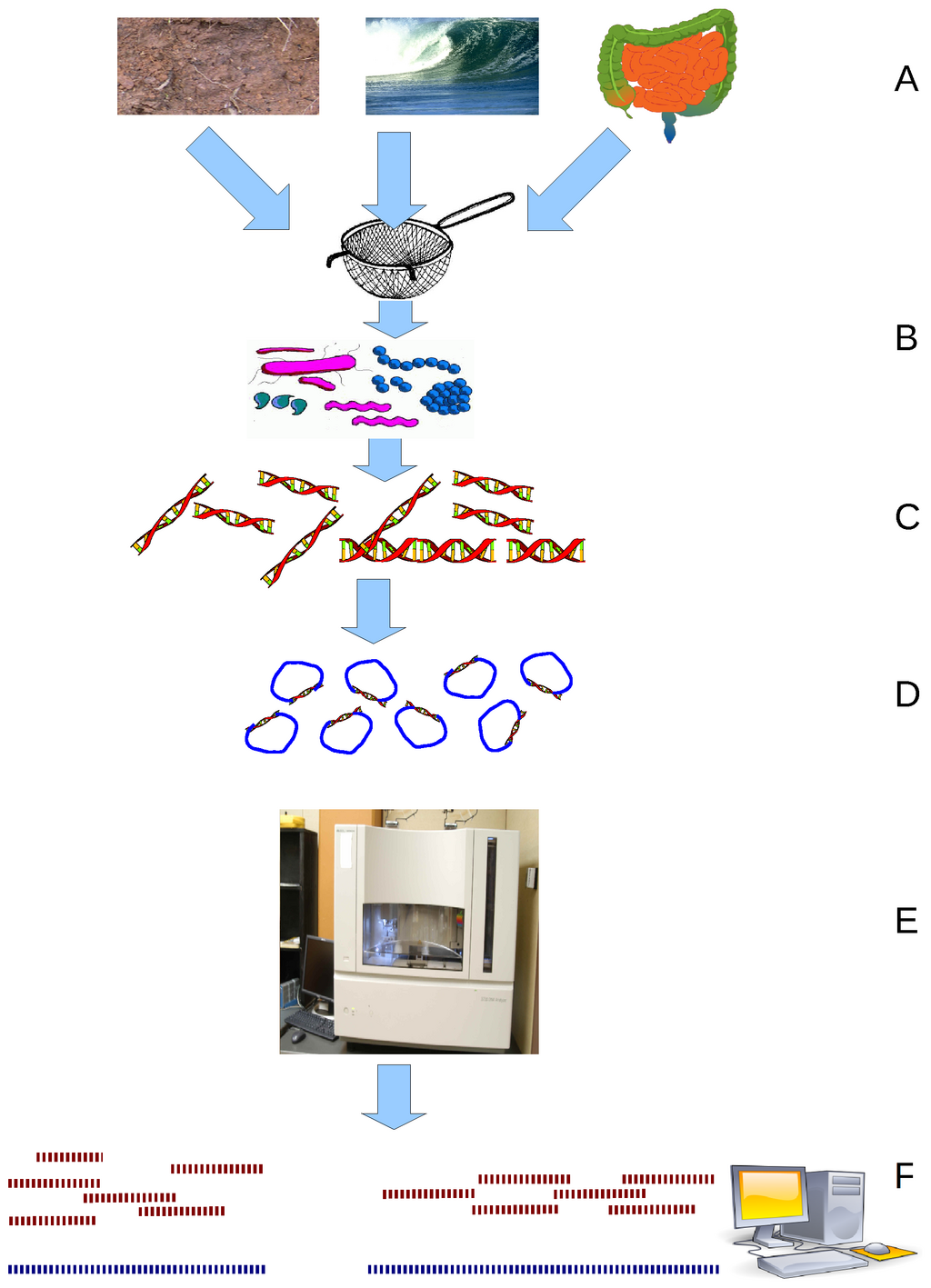 Environmental Shotgun Sequencing (ESS) is a key technique in metagenomics. (A) Sampling from habitat; (B) filtering particles, typically by size; (C) Lysis and DNA extraction; (D) cloning and library construction; (E) sequencing the clones; (F) sequence assembly into contigs and scaffolds. Main article: Metagenomics Metagenomics is the study of metagenomes, genetic material recovered directly from environmental samples. The broad field may also be referred to as environmental genomics, ecogenomics or community genomics. While traditional microbiology and microbial genome sequencing rely upon cultivated clonal cultures, early environmental gene sequencing cloned specific genes (often the 16S rRNA gene) to produce a profile of diversity in a natural sample. Such work revealed that the vast majority of microbial biodiversity had been missed by cultivation-based methods.[79] Recent studies use "shotgun" Sanger sequencing or massively parallel pyrosequencing to get largely unbiased samples of all genes from all the members of the sampled communities.[80] Because of its power to reveal the previously hidden diversity of microscopic life, metagenomics offers a powerful lens for viewing the microbial world that has the potential to revolutionize understanding of the entire living world.[81][82] Model systems Viruses and bacteriophages Bacteriophages have played and continue to play a key role in bacterial genetics and molecular biology. Historically, they were used to define gene structure and gene regulation. Also the first genome to be sequenced was a bacteriophage. However, bacteriophage research did not lead the genomics revolution, which is clearly dominated by bacterial genomics. Only very recently has the study of bacteriophage genomes become prominent, thereby enabling researchers to understand the mechanisms underlying phage evolution. Bacteriophage genome sequences can be obtained through direct sequencing of isolated bacteriophages, but can also be derived as part of microbial genomes. Analysis of bacterial genomes has shown that a substantial amount of microbial DNA consists of prophage sequences and prophage-like elements.[83] A detailed database mining of these sequences offers insights into the role of prophages in shaping the bacterial genome: Overall, this method verified many known bacteriophage groups, making this a useful tool for predicting the relationships of prophages from bacterial genomes.[84][85] Cyanobacteria At present there are 24 cyanobacteria for which a total genome sequence is available. 15 of these cyanobacteria come from the marine environment. These are six Prochlorococcus strains, seven marine Synechococcus strains, Trichodesmium erythraeum IMS101 and Crocosphaera watsonii WH8501. Several studies have demonstrated how these sequences could be used very successfully to infer important ecological and physiological characteristics of marine cyanobacteria. However, there are many more genome projects currently in progress, amongst those there are further Prochlorococcus and marine Synechococcus isolates, Acaryochloris and Prochloron, the N2-fixing filamentous cyanobacteria Nodularia spumigena, Lyngbya aestuarii and Lyngbya majuscula, as well as bacteriophages infecting marine cyanobaceria. Thus, the growing body of genome information can also be tapped in a more general way to address global problems by applying a comparative approach. Some new and exciting examples of progress in this field are the identification of genes for regulatory RNAs, insights into the evolutionary origin of photosynthesis, or estimation of the contribution of horizontal gene transfer to the genomes that have been analyzed.[86] |
研究分野 機能ゲノミクス 主な記事 機能ゲノミクス 機能ゲノミクスは分子生物学の一分野であり、ゲノム解読プロジェクトなどで得られる膨大なデータを利用して、遺伝子(およびタンパク質)の機能や相互作用 を記述しようとするものである。機能ゲノミクスは、DNA配列や構造といったゲノム情報の静的側面とは対照的に、遺伝子の転写、翻訳、タンパク質間相互作 用といった動的側面に焦点を当てている。機能ゲノミクスは、遺伝子、RNA転写産物、タンパク質産物のレベルで、DNAの機能に関する疑問に答えようとす るものである。機能ゲノミクス研究の主な特徴は、このような疑問に対するゲノムワイドなアプローチであり、一般的には、より伝統的な「遺伝子ごと」のアプ ローチではなく、ハイスループットメソッドを用いる。 ゲノミクスの主要な研究分野は、様々な生物のゲノムの塩基配列を決定することであるが、フルゲノムの知識は、主に様々な条件下での遺伝子発現パターンに関 係する機能ゲノミクスの分野の可能性を生み出した。ここで最も重要なツールはマイクロアレイとバイオインフォマティクスである。 構造ゲノミクス 主な記事 構造ゲノミクス 中西部構造ゲノミクスセンターが決定したタンパク質構造の例 構造ゲノミクスは、与えられたゲノムがコードする全てのタンパク質の3次元構造を記述しようとするものである[72][73]。このゲノムベースのアプ ローチにより、実験的アプローチとモデリングアプローチの組み合わせによるハイスループットな構造決定法が可能になる。構造ゲノミクスと従来の構造予測と の主な違いは、構造ゲノミクスでは、特定のタンパク質に焦点を当てるのではなく、ゲノムがコードするすべてのタンパク質の構造を決定しようとする点であ る。全ゲノム配列が利用できるようになったことで、構造予測は実験的アプローチとモデル化アプローチの組み合わせにより、より迅速に行えるようになった。 構造ゲノミクスでは、ゲノム配列を用いた実験的手法や、既知の構造を持つタンパク質との配列や構造の相同性に基づく、あるいは既知の構造とは相同性を持た ないタンパク質の化学的・物理的原理に基づくモデリングに基づくアプローチなど、構造決定のために多くのアプローチをとる。伝統的な構造生物学とは対照的 に、構造ゲノミクスによるタンパク質の構造決定は、タンパク質の機能が解明される前に行われることが多い(必ずしもそうではない)。このことは、構造バイ オインフォマティクス、すなわちタンパク質の3次元構造からタンパク質の機能を決定するという新たな課題を提起している[74]。 エピゲノミクス 主な記事 エピゲノミクス エピゲノミクスとは、エピゲノムとして知られる、細胞の遺伝物質上のエピジェネティックな修飾の完全な集合を研究することである[75]。エピジェネ ティックな修飾とは、細胞のDNAやヒストン上の可逆的な修飾であり、DNAの配列を変えることなく遺伝子発現に影響を与える(Russell 2010 p.475)。最も特徴的なエピジェネティック修飾の2つは、DNAのメチル化とヒストンの修飾である[76]。エピジェネティック修飾は、遺伝子の発現 と制御に重要な役割を果たし、分化/発生[77]や腫瘍形成など数多くの細胞プロセスに関与している[75]。グローバルレベルでのエピジェネティクスの 研究は、ゲノムハイスループットアッセイの適応により、最近になって可能となった[78]。 メタゲノミクス Environmental Shotgun Sequencing(ESS)は、メタゲノミクスにおける重要な技術である。(A)生息地からのサンプリング、(B)通常サイズ別に粒子をフィルター、 (C)溶解とDNA抽出、(D)クローニングとライブラリー構築、(E)クローンの配列決定、(F)コンティグとスキャフォールドへの配列アセンブル。 主な記事 メタゲノミクス メタゲノミクスとは、環境サンプルから直接回収した遺伝物質であるメタゲノムを研究することである。この広範な分野は、環境ゲノミクス、エコゲノミクス、 コミュニティゲノミクスとも呼ばれる。従来の微生物学や微生物ゲノムのシーケンシングが培養クローン培養に依存していたのに対し、初期の環境遺伝子シーケ ンシングは、特定の遺伝子(多くの場合16S rRNA遺伝子)をクローニングし、自然サンプル中の多様性のプロファイルを作成した。最近の研究では、「ショットガン」サンガーシーケンスや超並列パイ ロシーケンスを用いて、サンプリングされた生物群集の全メンバーから、ほぼ偏りのない全遺伝子のサンプルを得ることができる[80]。 モデル系 ウイルスとバクテリオファージ バクテリオファージは、細菌の遺伝学と分子生物学において重要な役割を担ってきたし、現在も担っている。歴史的には、バクテリオファージは遺伝子構造と遺 伝子制御を定義するために用いられた。また、塩基配列が決定された最初のゲノムはバクテリオファージであった。しかし、バクテリオファージの研究は、ゲノ ミクス革命をリードするものではなかった。ごく最近になって、バクテリオファージゲノムの研究が注目されるようになり、それによって研究者はファージの進 化の根底にあるメカニズムを理解することができるようになった。バクテリオファージのゲノム配列は、単離されたバクテリオファージの直接配列決定によって 得られるが、微生物ゲノムの一部として得られることもある。細菌ゲノムの解析により、かなりの量の微生物DNAがプロファージ配列やプロファージ様エレメ ントから構成されていることが示されている[83]。これらの配列の詳細なデータベースマイニングにより、細菌ゲノムの形成におけるプロファージの役割に ついての洞察が得られる: 全体として、この方法は多くの既知のバクテリオファージグループを検証し、細菌ゲノムからプロファージの関係を予測するための有用なツールとなった [84][85]。 シアノバクテリア 現在、全ゲノム配列が利用可能なシアノバクテリアは24種である。これらのシアノバクテリアのうち15種は海洋環境に由来する。これらは、6つのプロクロ ロコッカス株、7つの海洋性シネコッカス株、Trichodesmium erythraeum IMS101、Crocosphaera watsonii WH8501である。いくつかの研究は、これらの配列が海洋シアノバクテリアの重要な生態学的・生理学的特性を推測するのに非常にうまく利用できることを 示している。しかし、現在進行中のゲノムプロジェクトは、さらに多くのプロクロロコッカスや海洋性シネココッカス、アカリオクロリスやプロクロロン、N2 固定糸状藍藻ノデュラリア・スプミゲナ、リングビア・アエストゥアリイ、リングビア・マジスキュラ、そして海洋性藍藻に感染するバクテリオファージなどで ある。このように、増え続けるゲノム情報は、比較アプローチを適用することで、世界的な問題に対処するためのより一般的な方法で活用することもできる。こ の分野における進歩の新しいエキサイティングな例としては、制御RNAの遺伝子の同定、光合成の進化的起源に関する洞察、あるいは解析されたゲノムにおけ る水平遺伝子移動の寄与の推定などがある[86]。 |
Applications Schematic karyogram of a human, providing a simplified overview of the human genome. It is a graphical representation of the idealized human diploid karyotype, with annotated bands and sub-bands. It shows dark and white regions on G banding. Each row is vertically aligned at centromere level. It shows 22 homologous autosomal chromosome pairs, both the female (XX) and male (XY) versions of the two sex chromosomes, as well as the mitochondrial genome (at bottom left). Further information: Karyotype Genomics has provided applications in many fields, including medicine, biotechnology, anthropology and other social sciences.[44] Genomic medicine Next-generation genomic technologies allow clinicians and biomedical researchers to drastically increase the amount of genomic data collected on large study populations.[87] When combined with new informatics approaches that integrate many kinds of data with genomic data in disease research, this allows researchers to better understand the genetic bases of drug response and disease.[88][89] Early efforts to apply the genome to medicine included those by a Stanford team led by Euan Ashley who developed the first tools for the medical interpretation of a human genome.[90][91][92] The Genomes2People research program at Brigham and Women’s Hospital, Broad Institute and Harvard Medical School was established in 2012 to conduct empirical research in translating genomics into health. Brigham and Women's Hospital opened a Preventive Genomics Clinic in August 2019, with Massachusetts General Hospital following a month later.[93][94] The All of Us research program aims to collect genome sequence data from 1 million participants to become a critical component of the precision medicine research platform.[95] Synthetic biology and bioengineering The growth of genomic knowledge has enabled increasingly sophisticated applications of synthetic biology.[96] In 2010 researchers at the J. Craig Venter Institute announced the creation of a partially synthetic species of bacterium, Mycoplasma laboratorium, derived from the genome of Mycoplasma genitalium.[97] Population and conservation genomics Population genomics has developed as a popular field of research, where genomic sequencing methods are used to conduct large-scale comparisons of DNA sequences among populations - beyond the limits of genetic markers such as short-range PCR products or microsatellites traditionally used in population genetics. Population genomics studies genome-wide effects to improve our understanding of microevolution so that we may learn the phylogenetic history and demography of a population.[98] Population genomic methods are used for many different fields including evolutionary biology, ecology, biogeography, conservation biology and fisheries management. Similarly, landscape genomics has developed from landscape genetics to use genomic methods to identify relationships between patterns of environmental and genetic variation. Conservationists can use the information gathered by genomic sequencing in order to better evaluate genetic factors key to species conservation, such as the genetic diversity of a population or whether an individual is heterozygous for a recessive inherited genetic disorder.[99] By using genomic data to evaluate the effects of evolutionary processes and to detect patterns in variation throughout a given population, conservationists can formulate plans to aid a given species without as many variables left unknown as those unaddressed by standard genetic approaches.[100] |
アプリケーション ヒトゲノムの概要を簡略化したヒトの核型図。理想化されたヒトの二倍体核型を図式化したもので、バンドとサブバンドに注釈がついている。Gバンド上の濃い 領域と白い領域を示している。各行はセントロメアのレベルで縦に並んでいる。22本の常染色体の相同組、2本の性染色体の女性(XX)版と男性(XY) 版、およびミトコンドリアゲノム(左下)が示されている。 さらに詳しい情報はこちら: 核型 ゲノミクスは、医学、バイオテクノロジー、人類学、その他の社会科学など、多くの分野で応用されている[44]。 ゲノム医学 次世代ゲノム技術により、臨床医や生物医学研究者は大規模な研究集団について収集したゲノムデータの量を飛躍的に増やすことができる[87]。疾患研究に おいて、多くの種類のデータをゲノムデータと統合する新しいインフォマティクスのアプローチと組み合わせることで、研究者は薬物反応や疾患の遺伝的基盤を より深く理解することができる。 [ゲノムを医学に応用する初期の取り組みとしては、ヒトゲノムを医学的に解釈するための最初のツールを開発したEuan Ashley氏率いるスタンフォード大学のチームによるものがある[90][91][92]。ブリガム・アンド・ウィメンズ病院、ブロード研究所、ハー バード大学医学部のGenomes2People研究プログラムは、ゲノミクスを健康に応用するための実証的研究を行うために2012年に設立された。ブ リガム・アンド・ウィメンズ病院は2019年8月に予防ゲノミクスクリニックを開設し、その1ヵ月後にはマサチューセッツ総合病院も開設した[93] [94]。All of Us研究プログラムは、100万人の参加者からゲノム配列データを収集し、精密医療研究プラットフォームの重要な構成要素となることを目指している [95]。 合成生物学と生物工学 2010年、J.クレイグ・ベンター研究所の研究者たちは、マイコプラズマ・ジェニタリウムのゲノムから、部分的に合成された細菌種、マイコプラズマ・ラーバトリウムの創出を発表した[97]。 集団ゲノミクスと保全ゲノミクス 集団ゲノミクスは一般的な研究分野として発展しており、ゲノム配列決定法を用いて、集団遺伝学で伝統的に用いられてきた短距離PCR産物やマイクロサテラ イトなどの遺伝マーカーの限界を超えた、集団間のDNA配列の大規模な比較が行われている。集団ゲノミクスでは、ゲノムワイドな影響を研究し、ミクロ進化 の理解を深めることで、集団の系統発生史や人口動態を知ることができる。同様に、ランドスケープ・ゲノミクスは、ランドスケープ遺伝学から発展し、環境と 遺伝的変異のパターン間の関係を特定するためにゲノム手法を用いるようになった。 保全活動家は、個体群の遺伝的多様性や劣性遺伝性疾患に対するヘテロ接合の有無など、種の保全にとって重要な遺伝的要因をより適切に評価するために、ゲノ ム配列決定によって収集された情報を利用することができる[99]。進化のプロセスの影響を評価し、特定の個体群全体の変異のパターンを検出するためにゲ ノムデータを使用することにより、保全活動家は、標準的な遺伝学的アプローチでは対処できないような多くの変数を未知のままにしておくことなく、特定の種 を支援するための計画を策定することができる[100]。 |
| Hi-C (genomic analysis technique) Cognitive genomics Computational genomics Epigenomics Functional genomics GeneCalling, an mRNA profiling technology Genomics of domestication Genetics in fiction Glycomics Immunomics Metagenomics Pathogenomics Personal genomics Proteomics Transcriptomics Venomics Psychogenomics Whole genome sequencing Thomas Roderick |
Hi-C(ゲノム解析技術) 認知ゲノミクス 計算ゲノム学 エピゲノミクス 機能ゲノミクス GeneCalling(mRNAプロファイリング技術 家畜化のゲノミクス フィクションの中の遺伝学 グリコミクス イムノミクス メタゲノミクス 病理ゲノミクス パーソナルゲノミクス プロテオミクス トランスクリプトミクス ベノミクス サイコゲノミクス 全ゲノムシーケンス トーマス・ロデリック |
| https://en.wikipedia.org/wiki/Genomics |
|
●現代の骨相学:ゲノムサイエンス
東京大学の医科学研究所のヒトゲノム解析センター (Human Genome Center)は次のように その学問の目的がかかれてある(2021年3月19日採集)。
"The purpose of human genome research is to contribute to our human society through the diagnosis, prevention, and development of treatment methods for cancer, infectious diseases and other difficult diseases. The mission of the center is to promote this human genome research, greatly develop genomic medicine, and establish a new genomic medicine in the Society 5.0 era, and simultaneously build their foundations. To that end, we promote genomic medical science, medical informatics, artificial intelligence research for clinical translation of genomic information, and research on ethical, legal, and social issues. In addition, we operate a supercomputer system SHIROKANE for all researchers in life science regardless of academic or non-academic institutions/companies, and at the same time, provide seminars such as technical guidance course for users. We also accept researchers aiming at genomic research from all over the world and provide intensive courses for training." https://www.ims.u-tokyo.ac.jp/imsut/en/lab/hgclink/index.html
「ヒトゲノム研究の目的は、がんや感染症などの難治
性疾患の診断、予防、治療法の開発を通じて、人類社会に貢献することである。当センターの使命は、このヒトゲノム研究を推進し、ゲノム医療を飛躍的に発展
させ、Society
5.0時代の新しいゲノム医療を確立することであり、同時にその基盤を構築することである。そのため、ゲノム医療科学、医療情報学、ゲノム情報の臨床応用
に向けた人工知能研究、倫理・法・社会問題研究を推進している。また、学術・非学術機関・企業を問わず、生命科学の研究者向けにスーパーコンピュータシス
テム「SHIROKANE」を運用し、同時に利用者向け技術講習会などのセミナーも実施している。また、ゲノム研究を目指す世界中の研究者を受け入れ、集
中的なトレーニングコースを提供している」
★DNA Myth
リンク
文献
その他の情報

