
アルツハイマー病の生化学
Biochemistry of Alzheimer's disease
☆ 認知症の最も一般的な原因であるアルツハイマー病の生化学的メカニズムは、まだあまりよく理解されていない。アルツハイマー病(AD)はプロテオパシー、 すなわち、異常に折り畳まれたアミロイドβ(Aβ)タンパク質の脳内蓄積によるタンパク質のミスフォールディング病として同定されている。 [1] アミロイドβは、膜貫通タンパク質であるアミロイドβ前駆体タンパク質(APP)の異常なタンパク質分解副産物である短いペプチドであり、その機能は不明 であるが、神経細胞の発達に関与していると考えられている[2]。プレセニリンは、APPのプロセシングと分解に関与するタンパク質分解複合体の構成要素 である[3][4]。
★ このプロセスは、アミロイドカスケード仮説と呼ばれている()。
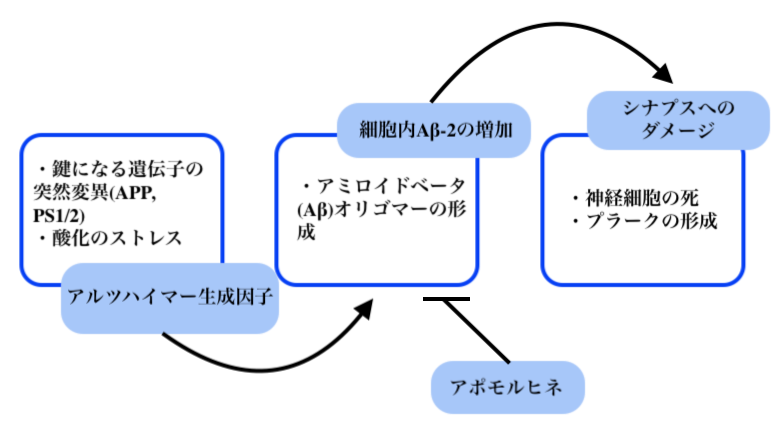
アルツハイマー病におけるアポモルヒネの役割を示すフローチャート.
| The biochemistry of Alzheimer's disease,
the most common cause of dementia, is not yet very well understood.
Alzheimer's disease (AD) has been identified as a proteopathy: a
protein misfolding disease due to the accumulation of abnormally folded
amyloid beta (Aβ) protein in the brain.[1] Amyloid beta is a short
peptide that is an abnormal proteolytic byproduct of the transmembrane
protein amyloid-beta precursor protein (APP), whose function is unclear
but thought to be involved in neuronal development.[2] The presenilins
are components of proteolytic complex involved in APP processing and
degradation.[3][4] Amyloid beta monomers are soluble and contain short regions of beta sheet and polyproline II helix secondary structures in solution,[5] though they are largely alpha helical in membranes;[6] however, at sufficiently high concentration, they undergo a dramatic conformational change to form a beta sheet-rich tertiary structure that aggregates to form amyloid fibrils.[7] These fibrils and oligomeric forms of Aβ deposit outside neurons in formations known as senile plaques. There are different types of plaques, including the diffuse, compact, cored or neuritic plaque types, as well as Aβ deposits in the walls of small blood vessel walls in the brain called cerebral amyloid angiopathy.[8][9] AD is also considered a tauopathy due to abnormal aggregation of the tau protein, a microtubule-associated protein expressed in neurons that normally acts to stabilize microtubules in the cell cytoskeleton. Like most microtubule-associated proteins, tau is normally regulated by phosphorylation; however, in Alzheimer's disease, hyperphosphorylated tau accumulates as paired helical filaments[10] that in turn aggregate into masses inside nerve cell bodies known as neurofibrillary tangles and as dystrophic neurites associated with amyloid plaques. Although little is known about the process of filament assembly, depletion of a prolyl isomerase protein in the parvulin family has been shown to accelerate the accumulation of abnormal tau.[11][12] Neuroinflammation is also involved in the complex cascade leading to AD pathology and symptoms. Considerable pathological and clinical evidence documents immunological changes associated with AD, including increased pro-inflammatory cytokine concentrations in the blood and cerebrospinal fluid.[13][14] Whether these changes may be a cause or consequence of AD remains to be fully understood, but inflammation within the brain, including increased reactivity of the resident microglia towards amyloid deposits, has been implicated in the pathogenesis and progression of AD.[15] Much of the known biochemistry of Alzheimer's disease has been deciphered through research using experimental models of Alzheimer's disease. |
認知症の最も一般的な原因であるアルツハイマー病の生化学的メカニズム
は、まだあまりよく理解されていない。アルツハイマー病(AD)はプロテオパシー、すなわち、異常に折り畳まれたアミロイドβ(Aβ)タンパク質の脳内蓄
積によるタンパク質のミスフォールディング病として同定されている。 [1]
アミロイドβは、膜貫通タンパク質であるアミロイドβ前駆体タンパク質(APP)の異常なタンパク質分解副産物である短いペプチドであり、その機能は不明
であるが、神経細胞の発達に関与していると考えられている[2]。プレセニリンは、APPのプロセシングと分解に関与するタンパク質分解複合体の構成要素
である[3][4]。 アミロイドβモノマーは可溶性で、溶液中ではβシートとポリプロリンIIらせんの二次構造の短い領域を含むが[5]、膜中では大部分がαらせん状である [6]。しかし、十分に高濃度になると、劇的な構造変化を起こしてβシートに富んだ三次構造を形成し、凝集してアミロイド線維を形成する[7]。プラーク には、びまん性プラーク、コンパクト・プラーク、コア型プラーク、神経斑型プラークなどの種類があり、脳アミロイド血管症と呼ばれる脳の細い血管壁への Aβ沈着もある[8][9]。 ADはまた、タウ蛋白質の異常凝集によるタウオパチーとも考えられている。タウ蛋白質は、神経細胞に発現する微小管関連蛋白質で、通常は細胞骨格の微小管 を安定化させる働きをする。ほとんどの微小管関連タンパク質と同様に、タウは通常リン酸化によって制御されている。しかし、アルツハイマー病では、高リン 酸化タウが一対のらせん状フィラメント[10]として蓄積し、それが神経原線維変化として知られる神経細胞体内の塊や、アミロイド斑に伴うジストロフィー 神経突起として凝集する。フィラメントが形成される過程についてはほとんど知られていないが、パルブリンファミリーに属するプロリルイソメラーゼタンパク 質の枯渇は、異常なタウの蓄積を促進することが示されている[11][12]。 神経炎症もまた、ADの病態や症状を引き起こす複雑なカスケードに関与している。かなりの病理学的および臨床的証拠が、血液中および脳脊髄液中の炎症性サ イトカイン濃度の上昇を含む、ADに関連した免疫学的変化を記録している。 [これらの変化がADの原因であるか結果であるかはまだ十分に理解されていないが、アミロイド沈着に対する常在ミクログリアの反応性の亢進を含む脳内の炎 症が、ADの病因と進行に関与していることが示唆されている。 |
| Neuropathology At a macroscopic level, AD is characterized by loss of neurons and synapses in the cerebral cortex and certain subcortical regions. This results in gross atrophy of the affected regions, including degeneration in the temporal lobe and parietal lobe, and parts of the frontal cortex and cingulate gyrus.[16] Both amyloid plaques and neurofibrillary tangles are clearly visible by microscopy in AD brains.[17] Plaques are dense, mostly insoluble deposits of protein and cellular material outside and around neurons. Tangles are insoluble twisted fibers that build up inside the nerve cell. Though many older people develop some plaques and tangles, the brains of AD patients have them to a much greater extent and in different brain locations.[18] |
神経病理学 巨視的レベルでは、ADは大脳皮質および特定の皮質下領域におけるニューロンとシナプスの喪失によって特徴づけられる。その結果、側頭葉と頭頂葉、前頭皮 質と帯状回の一部の変性など、罹患部位の肉眼的萎縮が生じる[16]。 ADの脳では、アミロイド斑と神経原線維のもつれの両方が顕微鏡ではっきりと確認できる[17]。もつれとは、神経細胞内に蓄積した不溶性のねじれた線維 のことである。多くの高齢者がプラークやもつれを生じているが、AD患者の脳には、より多くのプラークやもつれが存在し、脳の部位も異なる[18]。 |
| Biochemical characteristics Fundamental to the understanding of Alzheimer's disease is the biochemical events that leads to accumulation of the amyloid-beta plaques and tau-protein tangles. A delicate balance of the enzymes secretases regulate the amyloid-beta accumulation. Recently, a link between cholinergic neuronal activity and the activity of alpha-secretase has been highlighted,[19] which can discourage amyloid-beta proteins deposition in brain of patients with Alzheimer's disease. Alzheimer's disease has been identified as a protein misfolding disease, or proteopathy, due to the accumulation of abnormally folded amyloid-beta proteins in the brains of AD patients.[1] Abnormal amyloid-beta accumulation can first be detected using cerebrospinal fluid analysis and later using positron emission tomography (PET).[20] Although AD shares pathophysiological mechanisms with prion diseases, it is not transmissible in the wild, as prion diseases are.[21] Any transmissibility that it may have is limited solely to extremely rare iatrogenic events from donor-derived therapies that are no longer used.[22] Amyloid-beta, also written Aβ, is a short peptide that is a proteolytic byproduct of the transmembrane protein amyloid precursor protein (APP), whose function is unclear but thought to be involved in neuronal development. The presenilins are components of a proteolytic complex involved in APP processing and degradation.[4] Although amyloid beta monomers are harmless, they undergo a dramatic conformational change at sufficiently high concentration to form a beta sheet-rich tertiary structure that aggregates to form amyloid fibrils[7] that deposit outside neurons in dense formations known as senile plaques or neuritic plaques, in less dense aggregates as diffuse plaques, and sometimes in the walls of small blood vessels in the brain in a process called amyloid angiopathy or congophilic angiopathy. AD is also considered a tauopathy due to abnormal aggregation of the tau protein, a microtubule-associated protein expressed in neurons that normally acts to stabilize microtubules in the cell cytoskeleton. Like most microtubule-associated proteins, tau is normally regulated by phosphorylation; however, in AD patients, hyperphosphorylated tau accumulates as paired helical filaments[10] that in turn aggregate into masses inside nerve cell bodies known as neurofibrillary tangles and as dystrophic neurites associated with amyloid plaques. Levels of the neurotransmitter acetylcholine (ACh) are reduced. Levels of other neurotransmitters serotonin, norepinephrine, and somatostatin are also often reduced. Replenishing the ACh by anti-cholinesterases is an approved mode of treatment by FDA. An alternative method of stimulating ACh receptors of M1-M3 types by synthetic agonists that have a slower rate of dissociation from the receptor has been proposed as next generation cholinomimetic in Alzheimer's disease[15]. |
生化学的特徴 アルツハイマー病を理解する上で基本的なことは、アミロイドβ斑とタウ蛋白質のもつれを蓄積させる生化学的事象である。アミロイドβの蓄積は、セクレター ゼという酵素の微妙なバランスによって制御されている。最近、コリン作動性神経細胞の活性とαセクレターゼの活性との関連が注目されており[19]、アル ツハイマー病患者の脳におけるアミロイドβタンパク質の沈着を抑制することができる。アルツハイマー病は、AD患者の脳における異常な折り畳まれたアミロ イドベータタンパク質の蓄積により、タンパク質のミスフォールディング病、またはプロテオパシーとして同定されている[1]。異常なアミロイドベータの蓄 積は、まず脳脊髄液分析を用いて、後にポジトロン断層法(PET)を用いて検出することができる[20]。 ADはプリオン病と病態生理学的メカニズムを共有しているが、プリオン病のように自然界で感染することはない[21]。 [22] Aβとも呼ばれるアミロイドβは、膜貫通タンパク質であるアミロイド前駆体タンパク質(APP)のタンパク質分解副産物である短いペプチドであり、その機 能は不明であるが、神経細胞の発達に関与していると考えられている。プレセニリンはAPPのプロセシングと分解に関与するタンパク質分解複合体の構成要素 である。 [4] アミロイドβ単量体は無害であるが、十分に高濃度になると劇的な構造変化を起こし、βシートに富んだ三次構造を形成し、これが凝集してアミロイド線維を形 成する[7]。アミロイド線維は、老人斑または神経斑として知られる高密度な形態で神経細胞の外側に沈着し、拡散斑としてそれほど高密度でない凝集体で沈 着し、時にはアミロイド血管症または鬱血性血管症と呼ばれる過程で脳の細い血管壁に沈着する。 ADはまた、タウ蛋白質の異常凝集によるタウオパチーとも考えられている。タウ蛋白質は神経細胞に発現する微小管関連蛋白質で、通常は細胞骨格の微小管を 安定化させる働きをする。ほとんどの微小管関連蛋白質と同様に、タウは通常リン酸化によって制御されている。しかし、AD患者では、高リン酸化タウが一対 のらせん状フィラメント[10]として蓄積し、それが神経原線維変化として知られる神経細胞体内の塊や、アミロイド斑に関連するジストロフィー神経突起と して凝集する。 神経伝達物質であるアセチルコリン(ACh)の濃度は低下する。他の神経伝達物質であるセロトニン、ノルエピネフリン、ソマトスタチンのレベルもしばしば 低下する。抗コリンエステラーゼ薬によってAChを補充することは、FDAによって承認された治療法である。M1-M3型のACh受容体を、受容体からの 解離速度が遅い合成作動薬によって刺激する別の方法が、アルツハイマー病における次世代のコリン模倣薬として提案されている[15]。 |
| Disease mechanisms While the gross histological features of AD in the brain have been well characterized, several different hypotheses have been advanced regarding the primary cause. Among the oldest hypotheses is the cholinergic hypothesis, which suggests that deficiency in cholinergic signaling initiates the progression of the disease.[23] Current theories establish that both misfolding tau protein inside the cell and aggregation of amyloid beta outside the cell initiates the cascade leading to AD pathology.[24][25] Newer potential hypotheses propose metabolic factors,[26] vascular disturbance,[27] and chronically elevated inflammation in the brain[28] as contributing factors to AD. The amyloid beta hypothesis of molecular initiation have become dominant among many researchers to date.[29] The amyloid and tau hypothesis are the most widely accepted. Tau hypothesis The hypothesis that tau is the primary causative factor has long been grounded in the observation that deposition of amyloid plaques does not correlate well with neuron loss.[30] A mechanism for neurotoxicity has been proposed based on the loss of microtubule-stabilizing tau protein that leads to the degradation of the cytoskeleton.[31] However, consensus has not been reached on whether tau hyperphosphorylation precedes or is caused by the formation of the abnormal helical filament aggregates.[32] Support for the tau hypothesis also derives from the existence of other diseases known as tauopathies in which the same protein is identifiably misfolded.[33] However, a majority of researchers support the alternative hypothesis that amyloid is the primary causative agent.[32] Amyloid hypothesis The amyloid hypothesis was discovered because the gene for the amyloid beta precursor APP is located on chromosome 21, and patients with trisomy 21 – better known as Down syndrome – who have an extra gene copy exhibit AD-like disorders by 40 years of age.[34][35] The amyloid hypothesis points to the cytotoxicity of mature aggregated amyloid fibrils, which are believed to be the toxic form of the protein responsible for disrupting the cell's calcium ion homeostasis and thus inducing apoptosis.[36] This hypothesis is supported by the observation that higher levels of a variant of the beta amyloid protein known to form fibrils faster in vitro correlate with earlier onset and greater cognitive impairment in mouse models[37] and with AD diagnosis in humans.[38] However, mechanisms for the induced calcium influx, or proposals for alternative cytotoxic mechanisms, by mature fibrils are not obvious.[clarification needed] 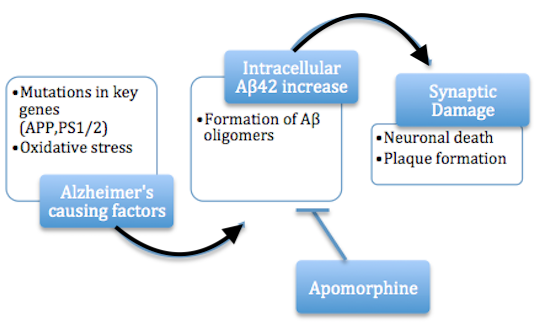 Flow chart depicting the role of apomorphine in Alzheimer's disease. A more recent variation of the amyloid hypothesis identifies the cytotoxic species as an intermediate misfolded form of amyloid beta, neither a soluble monomer nor a mature aggregated polymer but an oligomeric species, possibly toroidal or star-shaped with a central channel[39] that may induce apoptosis by physically piercing the cell membrane.[40] This ion channel hypothesis postulates that oligomers of soluble, non-fibrillar Aβ form membrane ion channels allowing unregulated calcium influx into neurons.[41] A related alternative suggests that a globular oligomer localized to dendritic processes and axons in neurons is the cytotoxic species.[42][43] The prefibrillar aggregates were shown to be able to disrupt the membrane.[44] The cytotoxic-fibril hypothesis presents a clear target for drug development: inhibit the fibrillization process. Much early development work on lead compounds has focused on this inhibition;[45][46][47] most are also reported to reduce neurotoxicity, but the toxic-oligomer theory would imply that prevention of oligomeric assembly is the more important process[48][49] [50] or that a better target lies upstream, for example in the inhibition of APP processing to amyloid beta.[51] For example, apomorphine was seen to significantly improve memory function through the increased successful completion of the Morris Water Maze.[48] Soluble intracellular (o)Aβ42 Two papers have shown that oligomeric (o)Aβ42 (a species of Aβ), in soluble intracellular form, acutely inhibits synaptic transmission, a pathophysiology that characterizes AD (in its early stages), by activating casein kinase 2.[52][53] Inflammatory hypothesis Converging evidence suggests that a sustained inflammatory response in the brain is a core modifying feature of AD pathology and may be a key modifying factor in AD pathogenesis.[54][55] The brains of AD patients exhibit several markers of increased inflammatory signaling.[56][57][58] The inflammatory hypothesis proposes that chronically elevated inflammation in the brain is a crucial component to the amyloid cascade[59] in the early phases of AD and magnifies disease severity in later stages of AD. Aβ is present in healthy brains and serves a vital physiological function in recovery from neuronal injury, protection from infection, and repair of the blood-brain barrier,[60] however it is unknown how Aβ production starts to exceed the clearance capacity of the brain and initiates AD progression. A possible explanation is that Aβ causes microglia, the resident immune cell of the brain, to become activated and secrete pro-inflammatory signaling molecules, called cytokines, which recruit other local microglia.[61] While acute microglial activation, as in response to injury, is beneficial and allows microglia to clear Aβ and other cellular debris via phagocytosis, chronically activated microglia exhibit decreased efficiency in Aβ clearance.[54] Despite this reduced AB clearance capacity, activated microglia continue to secrete pro-inflammatory cytokines like interleukins 1β and 6 (IL-6, IL-1β) and tumor necrosis factor-alpha (TNF-a), as well as reactive oxygen species which disrupt healthy synaptic functioning[62] and eventually cause neuronal death.[63] The loss of synaptic functioning and later neuronal death is responsible for the cognitive impairments and loss of volume in key brain regions which are associated with AD.[64] IL-1B, IL-6, and TNF-a cause further production of Aβ oligomers, as well as tau hyperphosphorylation, leading to continued microglia activation and creating a feed forward mechanism in which Aβ production is increased and Aβ clearance is decreased eventually causing the formation of Aβ plaques.[65][66] Historical cholinergic hypothesis The cholinergic hypothesis of AD development was first proposed in 1976 by Peter Davies and A.J.F Maloney.[67] It claimed that Alzheimer's begins as a deficiency in the production of acetylcholine, a vital neurotransmitter. Much early therapeutic research was based on this hypothesis, including restoration of the "cholinergic nuclei". The possibility of cell-replacement therapy was investigated on the basis of this hypothesis. All of the first-generation anti-Alzheimer's medications are based on this hypothesis and work to preserve acetylcholine by inhibiting acetylcholinesterases (enzymes that break down acetylcholine). These medications, though sometimes beneficial, have not led to a cure. In all cases, they have served to only treat symptoms of the disease and have neither halted nor reversed it. These results and other research have led to the conclusion that acetylcholine deficiencies may not be directly causal, but are a result of widespread brain tissue damage, damage so widespread that cell-replacement therapies are likely to be impractical. More recent findings center on the effects of the misfolded and aggregated proteins, amyloid beta and tau: tau protein abnormalities may initiate the disease cascade, then beta amyloid deposits progress the disease.[32] Glucose consumption The human brain is one of the most metabolically active organs in the body and metabolizes a large amount of glucose to produce cellular energy in the form of adenosine triphosphate (ATP).[68] Despite its high energy demands, the brain is relatively inflexible in its ability to utilize substrates for energy production and relies almost entirely on circulating glucose for its energy needs.[69] This dependence on glucose puts the brain at risk if the supply of glucose is interrupted, or if its ability to metabolize glucose becomes defective. If the brain is not able to produce ATP, synapses cannot be maintained and cells cannot function, ultimately leading to impaired cognition.[69] Imaging studies have shown decreased utilization of glucose in the brains of Alzheimer's disease patients early in the disease, before clinical signs of cognitive impairment occur. This decrease in glucose metabolism worsens as clinical symptoms develop and the disease progresses.[70][71] Studies have found a 17%-24% decline in cerebral glucose metabolism in patients with Alzheimer's disease, compared with age-matched controls.[72] Numerous imaging studies have since confirmed this observation. Abnormally low rates of cerebral glucose metabolism are found in a characteristic pattern in the Alzheimer's disease brain, particularly in the posterior cingulate, parietal, temporal, and prefrontal cortices. These brain regions are believed to control multiple aspects of memory and cognition. This metabolic pattern is reproducible and has even been proposed as a diagnostic tool for Alzheimer's disease. Moreover, diminished cerebral glucose metabolism (DCGM) correlates with plaque density and cognitive deficits in patients with more advanced disease.[72][73] Diminished cerebral glucose metabolism (DCGM) may not be solely an artifact of brain cell loss since it occurs in asymptomatic patients at risk for Alzheimer's disease, such as patients homozygous for the epsilon 4 variant of the apolipoprotein E gene (APOE4, a genetic risk factor for Alzheimer's disease), as well as in inherited forms of Alzheimer's disease.[74] Given that DCGM occurs before other clinical and pathological changes occur, it is unlikely to be due to the gross cell loss observed in Alzheimer's disease.[69] In imaging studies involving young adult APOE4 carriers, where there were no signs of cognitive impairment, diminished cerebral glucose metabolism (DCGM) was detected in the same areas of the brain as older subjects with Alzheimer's disease.[74] However, DCGM is not exclusive to APOE4 carriers. By the time Alzheimer's has been diagnosed, DCGM occurs in genotypes APOE3/E4, APOE3/E3, and APOE4/E4.[75] Thus, DCGM is a metabolic biomarker for the disease state.[76] Insulin signaling A connection has been established between Alzheimer's disease and diabetes during the past decade, as insulin resistance, which is a characteristic hallmark of diabetes, has also been observed in brains of subjects with Alzheimer's disease.[77] Neurotoxic oligomeric amyloid-β species decrease the expression of insulin receptors on the neuronal cell surface[78] and abolish neuronal insulin signaling.[77] It has been suggested that neuronal gangliosides, which take part in the formation of membrane lipid microdomains, facilitate amyloid-β-induced removal of the insulin receptors from the neuronal surface.[79] In Alzheimer's disease, oligomeric amyloid-β species trigger TNF-α signaling.[77] c-Jun N-terminal kinase activation by TNF-α in turn activates stress-related kinases and results in IRS-1 serine phosphorylation, which subsequently blocks downstream insulin signaling.[77][80][81] The resulting insulin resistance contributes to cognitive impairment. Consequently, increasing neuronal insulin sensitivity and signaling may constitute a novel therapeutic approach to treat Alzheimer's disease.[82][83] Oxidative stress Oxidative stress is emerging as a key factor in the pathogenesis of AD.[84] Reactive oxygen species (ROS) over-production is thought to play a critical role in the accumulation and deposition of amyloid beta in AD.[85] Brains of AD patients have elevated levels of oxidative DNA damage in both nuclear and mitochondrial DNA, but the mitochondrial DNA has approximately 10-fold higher levels than nuclear DNA.[86] Aged mitochondria may be the critical factor in the origin of neurodegeneration in AD.[85] Even individuals with mild cognitive impairment, the phase between normal aging and early dementia, have increased oxidative damage in their nuclear and mitochondrial brain DNA[87] (see Aging brain). Naturally occurring DNA double-strand breaks (DSBs) arise in human cells largely from single-strand breaks induced by various processes including the activity of reactive oxygen species, topoisomerases, and hydrolysis due to thermal fluctuations.[88] In neurons DSBs are induced by a type II topoisomerase as part of the physiologic process of memory formation.[89] DSBs are present in both neurons and astrocytes in the postmortem human hippocampus of AD patients at a higher level than in non-AD individuals.[90] AD is associated with an accumulation of DSBs in neurons and astrocytes in the hippocampus and frontal cortex from early stages onward.[91] DSBs are increased in the vicinity of amyloid plaques in the hippocampus, indicating a potential role for Aβ in DSB accumulation or vice versa.[90] The predominant mechanism for repairing DNA double-strand breaks is non-homologous end joining (NHEJ), a mechanism that utilizes the DNA-dependent protein kinase (DNA-PK) complex. The end joining activity and protein levels of DNA-PK catalytic subunit are significantly lower in AD brains than in normal brains.[92] Cholesterol hypothesis The cholesterol hypothesis is a combination of the amyloid hypothesis, tau hypothesis, and potentially the inflammatory hypothesis. Cholesterol was shown to be upstream of both amyloid and tau production.[93] The cholesterol is produced in the astrocytes and shipped to neurons where it activates amyloid production through a process called substrate presentation. The process required apoE. Cholesterol's regulation of Tau production is less well understood, but knocking out the cholesterol synthesis enzyme SREBP2 decreased Tau phosphorylation. [94] Innate immunity triggers cholesterol synthesis and cells take up the cholesterol.[95] Presumably a cell in the brain dies with old age and this triggers innate immunity. More studies are needed to directly tie the inflammatory hypothesis to cholesterol synthesis in the brain. Reelin hypothesis A 1994 study [96] showed that the isoprenoid changes in Alzheimer's disease differ from those occurring during normal aging and that this disease cannot, therefore, be regarded as a result of premature aging. During aging the human brain shows a progressive increase in levels of dolichol, a reduction in levels of ubiquinone, but relatively unchanged concentrations of cholesterol and dolichyl phosphate. In Alzheimer's disease, the situation is reversed with decreased levels of dolichol and increased levels of ubiquinone. The concentrations of dolichyl phosphate are also increased, while cholesterol remains unchanged. The increase in the sugar carrier dolichyl phosphate may reflect an increased rate of glycosylation in the diseased brain and the increase in the endogenous anti-oxidant ubiquinone an attempt to protect the brain from oxidative stress, for instance induced by lipid peroxidation.[96] Ropren, identified previously in Russia, is neuroprotective in a rat model of Alzheimer's disease.[97][98] A relatively recent hypothesis based mainly on rodent experiments links the onset of Alzheimer's disease to the hypofunction of the large extracellular protein reelin. A decrease of reelin in the human entorhinal cortex where the disease typically initiates is evident [99] while compensatory increase of reelin levels in other brain structures of the patients is also reported.[100] Of key importance, overexpression of reelin rescues the cognitive capacities of Alzheimer's disease model mice [101] and τ-protein overexpressing mice.[102] A recent circuit level model proposed a mechanism of how reelin depletion leads to the early deterioration of episodic memory thereby laying the theoretical foundation of the reelin hypothesis.[103] Large gene instability hypothesis A bioinformatics analysis in 2017[104] revealed that extremely large human genes are significantly over-expressed in brain and take part in the postsynaptic architecture. These genes are also highly enriched in cell adhesion Gene Ontology (GO) terms and often map to chromosomal fragile sites.[105] The majority of known Alzheimer's disease risk gene products including the amyloid precursor protein (APP) and gamma-secretase, as well as the APOE receptors and GWAS risk loci take part in similar cell adhesion mechanisms. It was concluded that dysfunction of cell and synaptic adhesion is central to Alzheimer's disease pathogenesis, and mutational instability of large synaptic adhesion genes may be the etiological trigger of neurotransmission disruption and synaptic loss in brain aging. As a typical example, this hypothesis explains the APOE risk locus of AD in context of signaling of its giant lipoprotein receptor, LRP1b which is a large tumor-suppressor gene with brain-specific expression and also maps to an unstable chromosomal fragile site. The large gene instability hypothesis puts the DNA damage mechanism at the center of Alzheimer's disease pathophysiology. |
疾患のメカニズム 脳におけるADの肉眼的組織学的特徴はよく特徴付けられているが、主な原因についてはいくつかの異なる仮説が提唱されてきた。最も古い仮説はコリン作動性 仮説であり、コリン作動性シグナル伝達の欠乏が疾患の進行を開始させることを示唆している[23]。現在の理論では、細胞内でのタウ蛋白のミスフォール ディングと細胞外でのアミロイドβの凝集の両方がAD病態に至るカスケードを開始させるとしている[24][25]。現在までに多くの研究者の間では、ア ミロイドベータ仮説が主流となっている[29]。アミロイド仮説とタウ仮説が最も広く受け入れられている。 タウ仮説 タウが主要な原因因子であるという仮説は、アミロイド斑の沈着が神経細胞の喪失とあまり相関しないという観察に基づいて長い間提唱されてきた[30]。細 胞骨格の分解につながる微小管安定化タウタンパク質の喪失に基づく神経毒性のメカニズムが提唱されてきた。 [31] しかしながら、タウの過リン酸化が異常ならせん状フィラメント凝集体の形成に先行するのか、あるいはそれによって引き起こされるのかについては、意見の一 致をみていない。 [32] タウ仮説に対する支持は、同じタンパク質が同定可能にミスフォールドするタウオパチーとして知られる他の疾患の存在にも由来する[33]。しかしながら、 研究者の大多数は、アミロイドが主要な原因物質であるという代替仮説を支持している[32]。 アミロイド仮説 アミロイド仮説が発見されたのは、アミロイドβ前駆体APPの遺伝子が21番染色体上にあり、21トリソミー(ダウン症候群としてよく知られている)患者 で遺伝子のコピーが余っている場合、40歳までにAD様の障害を示すからである。 [34][35]アミロイド仮説は、成熟凝集アミロイド線維の細胞毒性を指摘しており、この線維は細胞のカルシウムイオンのホメオスタシスを破壊し、アポ トーシスを誘導するタンパク質の毒性形態であると考えられている。 [36] この仮説は、in vitroでより速く線維を形成することが知られているβアミロイドタンパク質の変異体のレベルが高いほど、マウスモデル[37]での早期発症やより大き な認知障害と相関し、ヒトでのAD診断と相関するという観察によって支持されている[38]。しかしながら、成熟線維によるカルシウム流入誘導のメカニズ ムや、代替の細胞毒性メカニズムの提案は明らかではない。 アルツハイマー病におけるアポモルヒネの役割を示すフローチャート。 アミロイド仮説のより最近のバリエーションでは、細胞毒性種はアミロイドβの中間的なミスフォールディング型であり、可溶性の単量体でも成熟した凝集ポリ マーでもなく、オリゴマー型であり、おそらく中心部にチャネルを持つトロイダル型か星型であり[39]、細胞膜を物理的に貫通することによってアポトーシ スを誘導する可能性がある。 [40]。このイオンチャネル仮説は、可溶性で非線維性のAβのオリゴマーが膜イオンチャネルを形成し、ニューロンへの無秩序なカルシウム流入を可能にす ると仮定している[41]。関連する代替案は、ニューロンの樹状突起や軸索に局在する球状オリゴマーが細胞毒性種であることを示唆している[42] [43]。 細胞毒性線維仮説は、薬剤開発の明確なターゲットを提示する。リード化合物に関する初期の開発研究の多くは、この阻害に焦点が当てられている。[45] [46][47]多くは神経毒性を軽減することも報告されているが、毒性オリゴマー理論は、オリゴマー集合の防止がより重要なプロセスであること[48] [49][50]、あるいは、より良い標的が上流にあること、例えばAPPのアミロイドβへのプロセシングの阻害にあることを示唆している[51]。 可溶性細胞内(o)Aβ42 可溶性細胞内型オリゴマー(o)Aβ42(Aβの一種)が、カゼインキナーゼ2を活性化することによって、AD(の初期段階)を特徴づける病態生理である シナプス伝達を急性的に阻害することが、2つの論文で示されている[52][53]。 炎症仮説 AD患者の脳は、炎症シグナル伝達の亢進を示すいくつかのマーカーを示す[56][57][58]。炎症仮説では、脳内で慢性的に亢進する炎症は、ADの 初期段階ではアミロイドカスケード[59]の重要な構成要素であり、ADの後期段階では疾患の重症度を増大させると提唱している。Aβは健康な脳にも存在 し、神経細胞傷害からの回復、感染からの保護、血液脳関門の修復において重要な生理的機能を果たしている[60]が、Aβの産生が脳のクリアランス能力を どのようにして上回り始め、ADの進行を開始するのかは不明である。可能性のある説明としては、Aβによって脳の常在免疫細胞であるミクログリアが活性化 され、サイトカインと呼ばれる炎症性シグナル分子を分泌し、他の局所のミクログリアをリクルートするというものである[61]。傷害に反応したときのよう な急性のミクログリアの活性化は有益であり、ミクログリアが貪食作用によってAβや他の細胞残屑を除去することを可能にするが、慢性的に活性化されたミク ログリアは、Aβクリアランスの効率低下を示す。 [54] このようにABクリアランス能力が低下しているにもかかわらず、活性化ミクログリアは、インターロイキン1βおよび6(IL-6、IL-1β)、腫瘍壊死 因子α(TNF-a)などの炎症性サイトカインや、健全なシナプス機能を破壊し[62]、最終的には神経細胞死を引き起こす活性酸素種を分泌し続ける。 [63]シナプス機能の喪失とその後の神経細胞死は、ADに関連する脳の主要部位の認知障害と容積の減少の原因である[64]。IL-1B、IL-6、 TNF-aは、Aβオリゴマーのさらなる産生とタウの過剰リン酸化を引き起こし、ミクログリアの活性化を継続させ、Aβ産生が増加しAβクリアランスが減 少するフィードフォワード機構を作り出し、最終的にAβ斑の形成を引き起こす[65][66]。 歴史的コリン作動性仮説 AD発症のコリン作動性仮説は、1976年にPeter DaviesとA.J.F Maloneyによって初めて提唱された[67]。この仮説は、アルツハイマー病は重要な神経伝達物質であるアセチルコリンの産生不全から始まると主張し た。初期の治療研究の多くは、「コリン作動性核」の回復など、この仮説に基づいていた。この仮説に基づいて、細胞補充療法の可能性が研究された。第一世代 の抗アルツハイマー薬はすべてこの仮説に基づいており、アセチルコリンエステラーゼ(アセチルコリンを分解する酵素)を阻害することによってアセチルコリ ンを温存するように働いている。これらの薬は、時に有益ではあるが、治療には至っていない。いずれの場合も、病気の症状を治療するだけで、病気を食い止め ることも回復させることもできなかった。これらの結果や他の研究により、アセチルコリンの欠乏が直接の原因ではなく、脳組織の広範囲に及ぶ損傷の結果であ るという結論が導き出された。 より最近の知見では、ミスフォールドして凝集したタンパク質であるアミロイドβとタウの影響が中心となっている。タウタンパク質の異常が疾患カスケードを 開始し、その後βアミロイドの沈着が疾患を進行させる可能性がある[32]。 グルコースの消費 ヒトの脳は、体内で最も代謝活性の高い臓器の一つであり、アデノシン三リン酸(ATP)の形で細胞エネルギーを産生するために、大量のグルコースを代謝す る[68]。その高いエネルギー需要にもかかわらず、脳はエネルギー産生のための基質を利用する能力において比較的柔軟性に欠け、そのエネルギー必要量を ほぼ完全に循環グルコースに依存している[69]。このグルコースへの依存は、グルコースの供給が中断されたり、グルコースを代謝する能力に欠陥が生じた りした場合に、脳を危険にさらす。脳がATPを産生できなくなると、シナプスが維持できなくなり、細胞が機能しなくなるため、最終的に認知能力の低下につ ながる。 画像研究によると、アルツハイマー病患者の脳では、認知機能障害の臨床的徴候が現れる前、疾患の初期においてグルコースの利用が低下していることが示され ている。このグルコース代謝の低下は、臨床症状が発現し、疾患が進行するにつれて悪化する[70][71]。研究により、アルツハイマー病患者では、年齢 をマッチさせた対照群と比較して、大脳グルコース代謝が17~24%低下していることが明らかにされている[72]。 その後、多数の画像研究によりこの観察が確認されている。 脳内グルコース代謝の異常な低下は、アルツハイマー病の脳、特に後帯状皮質、頭頂皮質、側頭皮質、前頭前皮質において特徴的なパターンで認められる。これ らの脳領域は記憶と認知の多面的な側面を制御していると考えられている。この代謝パターンは再現可能であり、アルツハイマー病の診断ツールとしても提案さ れている。さらに、脳糖代謝の低下(DCGM)は、より進行した疾患におけるプラーク密度および認知障害と相関する[72][73]。 脳グルコース代謝の低下(DCGM)は、アルツハイマー病の遺伝的危険因子であるアポリポ蛋白E遺伝子のε4変異体(APOE4)のホモ接合体のようなア ルツハイマー病の危険性のある無症候性の患者や遺伝性のアルツハイマー病で起こるため、脳細胞の減少によるものだけではない可能性がある。 [74] DCGMは、他の臨床的および病理学的変化が起こる前に起こることから、アルツハイマー病で観察される総体的な細胞消失によるものとは考えにくい [69]。 認知機能障害の徴候がない若年成人のAPOE4キャリアの画像研究において、減少した脳糖代謝(DCGM)が、アルツハイマー病の高齢被験者と同じ脳の領 域で検出された[74]。アルツハイマー病が診断されるまでに、DCGMはAPOE3/E4、APOE3/E3、APOE4/E4の遺伝子型に発現する [75]。したがって、DCGMは疾患状態の代謝バイオマーカーである[76]。 インスリンシグナル伝達 糖尿病の特徴的な徴候であるインスリン抵抗性がアルツハイマー病の被験者の脳でも観察されたことから、過去10年間にアルツハイマー病と糖尿病の間に関連 性が確立された[77]。神経毒性オリゴマーアミロイドβ種は、神経細胞表面のインスリン受容体の発現を減少させ[78]、神経細胞のインスリンシグナル 伝達を消失させる。 [77] 膜脂質ミクロドメインの形成に関与する神経細胞ガングリオシドが、アミロイドβによって誘導される神経細胞表面からのインスリン受容体の除去を促進するこ とが示唆されている。 [77]。TNF-αによるc-Jun N末端キナーゼの活性化は、ストレス関連キナーゼを活性化し、IRS-1のセリンリン酸化をもたらし、その結果、下流のインスリンシグナル伝達が阻害され る[77][80][81]。 結果として生じるインスリン抵抗性は、認知機能障害の一因となる。その結果、神経細胞のインスリン感受性とシグナル伝達を高めることが、アルツハイマー病 を治療するための新たな治療法となる可能性がある[82][83]。 酸化ストレス 酸化ストレスは、ADの病因における重要な因子として浮上している。[84] 活性酸素種(ROS)の過剰産生は、ADにおけるアミロイドβの蓄積と沈着において重要な役割を果たすと考えられている[85] AD患者の脳では、核DNAとミトコンドリアDNAの両方において酸化的DNA損傷のレベルが上昇しているが、ミトコンドリアDNAは核DNAの約10倍 高いレベルである。 [正常な老化と早期認知症の中間に位置する軽度認知障害でさえ、核およびミトコンドリアの脳DNAにおける酸化的損傷が増加している[87](脳の老化を 参照)。自然発生するDNAの二本鎖切断(DSB)は、ヒトの細胞では、活性酸素種、トポイソメラーゼの活性、熱揺らぎによる加水分解など、様々な過程に よって誘導される一本鎖切断から主に生じる[88]。神経細胞では、DSBは、記憶形成の生理学的過程の一部としてII型トポイソメラーゼによって誘導さ れる[89]。 [90]ADは、初期段階から海馬と前頭皮質の神経細胞とアストロサイトにおけるDSBの蓄積と関連している[91]。DSBは海馬のアミロイド斑の近傍 で増加しており、DSBの蓄積におけるAβの役割、あるいはその逆の役割の可能性を示している。 [90] DNAの二本鎖切断を修復する主なメカニズムは非相同末端接合(NHEJ)であり、DNA依存性プロテインキナーゼ(DNA-PK)複合体を利用するメカ ニズムである。DNA-PK触媒サブユニットの末端接合活性とタンパク質レベルは、ADの脳では正常の脳よりも有意に低い[92]。 コレステロール仮説 コレステロール仮説は、アミロイド仮説、タウ仮説、そして潜在的には炎症仮説を組み合わせたものである。コレステロールはアミロイドとタウの両方の産生の 上流にあることが示された[93]。コレステロールはアストロサイトで産生され、ニューロンに輸送され、そこで基質提示と呼ばれるプロセスを通じてアミロ イド産生を活性化する。このプロセスにはアポEが必要であった。コレステロールによるタウ産生の制御についてはあまりよく分かっていないが、コレステロー ル合成酵素SREBP2をノックアウトすると、タウのリン酸化が減少した。[94]自然免疫はコレステロール合成を誘発し、細胞はコレステロールを取り込 む[95]。炎症仮説と脳内のコレステロール合成を直接結びつけるには、さらなる研究が必要である。 リーリン仮説 1994年の研究[96]により、アルツハイマー病におけるイソプレノイドの変化は、正常な老化の過程で起こる変化とは異なることが示され、したがってこ の病気は早期老化の結果とは考えられないことが示された。加齢に伴い、ヒトの脳はドリコールの濃度が徐々に増加し、ユビキノンの濃度が減少するが、コレス テロールとドリチルリン酸の濃度は比較的変化しない。アルツハイマー病では、ドリコールの濃度が低下し、ユビキノンの濃度が上昇する。ドリチルリン酸の濃 度も上昇し、コレステロールは変化しない。糖キャリアであるドリチルリン酸の増加は、病的脳におけるグリコシル化速度の増加を反映していると考えられ、内 因性抗酸化物質であるユビキノンの増加は、例えば脂質過酸化によって誘導される酸化ストレスから脳を保護しようとする試みであると考えられる[96]。ロ シアで以前に同定されたロプレンは、アルツハイマー病のラットモデルにおいて神経保護作用を示す[97][98]。 主にげっ歯類の実験に基づく比較的最近の仮説は、アルツハイマー病の発症を大きな細胞外タンパク質であるリーリンの機能低下と関連付けるものである。典型 的な発症部位であるヒトの嗅内皮質におけるリーリンの減少は明らかであり [99] 、一方で患者の他の脳構造におけるリーリンレベルの代償的増加も報告されている。 [100] 特に重要なのは、リーリンを過剰発現させると、アルツハイマー病モデルマウス [101] やτタンパク質過剰発現マウスの認知能力が回復することである。 大規模遺伝子不安定性仮説 2017年のバイオインフォマティクス解析[104]により、非常に大きなヒト遺伝子が脳内で著しく過剰発現しており、シナプス後アーキテクチャに関与し ていることが明らかになった。これらの遺伝子はまた、細胞接着のGene Ontology(GO)用語に非常に富んでおり、しばしば染色体脆弱部位にマップされる[105]。アミロイド前駆体タンパク質(APP)やγセクレ ターゼ、APOE受容体やGWASリスク遺伝子座を含む既知のアルツハイマー病リスク遺伝子産物の大部分は、同様の細胞接着メカニズムに関与している。細 胞接着とシナプス接着の機能不全がアルツハイマー病の病因の中心であり、大きなシナプス接着遺伝子の変異不安定性が、脳の老化における神経伝達の混乱とシ ナプス喪失の病因論的引き金である可能性があると結論づけられた。典型的な例として、この仮説は、ADのAPOEリスク遺伝子座を、脳特異的発現を持つ大 きな腫瘍抑制遺伝子であり、不安定な染色体脆弱部位にもマップされる巨大リポタンパク質受容体LRP1bのシグナル伝達との関連で説明する。大規模遺伝子 不安定性仮説は、DNA損傷機構をアルツハイマー病病態生理の中心に据えている。 |
| https://en.wikipedia.org/wiki/Biochemistry_of_Alzheimer%27s_disease |
|
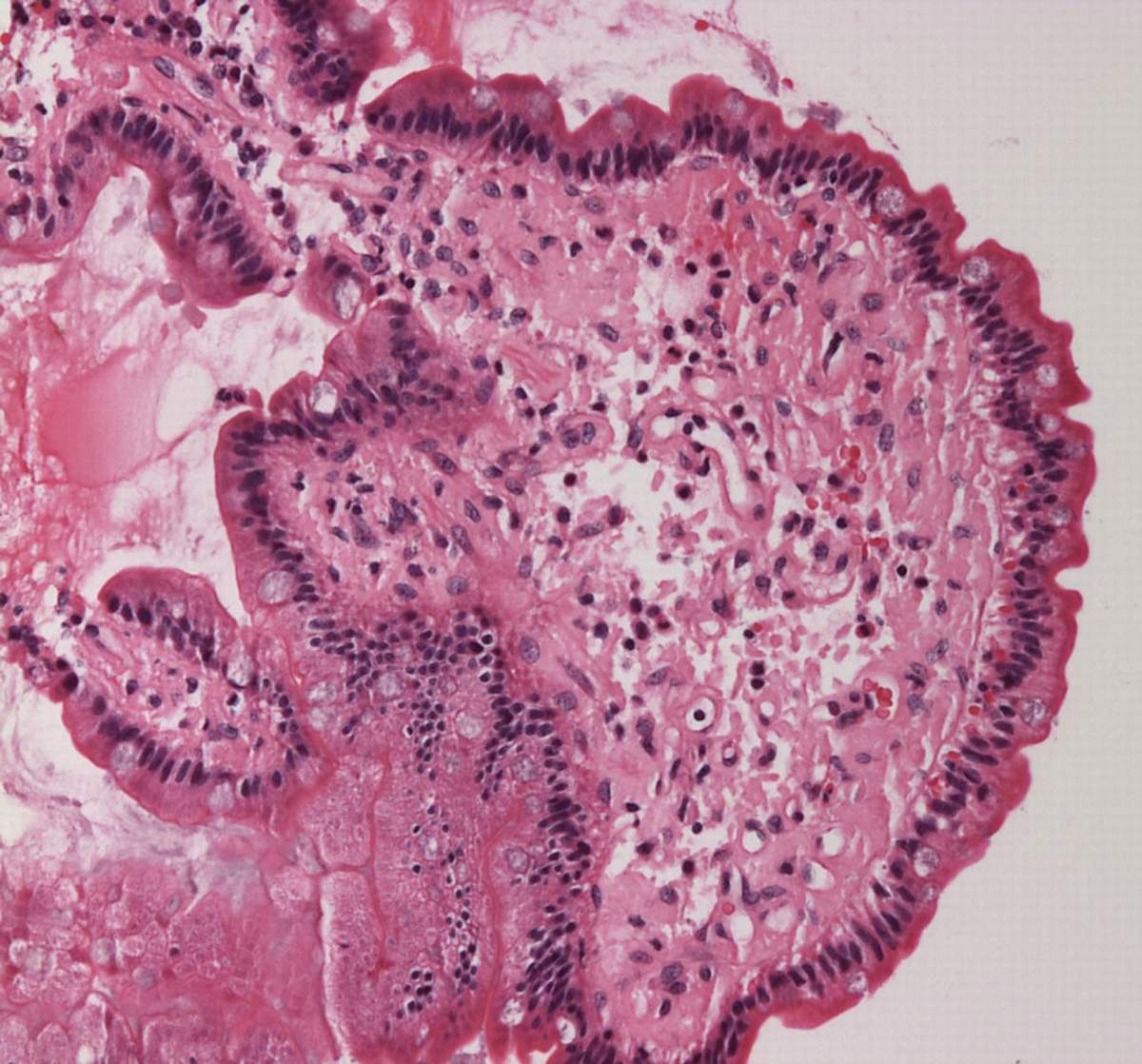 Micrograph showing amyloid deposits (pink) in small bowel. Duodenum with amyloid deposition in lamina propria. Amyloid shows up as homogeneous pink material in lamina propria and around blood vessels. 20× magnification. |
小腸のアミロイド沈着(ピンク色)を示す顕微鏡写真。アミロイドが固有層に沈着している十二指腸。アミロイドは固有層と血管周囲に均一なピンク色の物質と して認められる。倍率20倍。 |
| Amyloids are
aggregates of proteins characterised by a fibrillar morphology of
typically 7–13 nm in diameter, a β-sheet secondary structure (known as
cross-β) and ability to be stained by particular dyes, such as Congo
red.[1] In the human body, amyloids have been linked to the development
of various diseases.[2] Pathogenic amyloids form when previously
healthy proteins lose their normal structure and physiological
functions (misfolding) and form fibrous deposits within and around
cells. These protein misfolding and deposition processes disrupt the
healthy function of tissues and organs. Such amyloids have been associated with (but not necessarily as the cause of) more than 50[2][3] human diseases, known as amyloidosis, and may play a role in some neurodegenerative diseases.[2][4] Some of these diseases are mainly sporadic and only a few cases are familial. Others are only familial. Some result from medical treatment. Prions are an infectious form of amyloids that can act as a template to convert other non-infectious forms.[5] Amyloids may also have normal biological functions; for example, in the formation of fimbriae in some genera of bacteria, transmission of epigenetic traits in fungi, as well as pigment deposition and hormone release in humans.[6] Amyloids have been known to arise from many different proteins.[2][7] These polypeptide chains generally form β-sheet structures that aggregate into long fibers; however, identical polypeptides can fold into multiple distinct amyloid conformations.[8] The diversity of the conformations may have led to different forms of the prion diseases.[6] An unusual secondary structure named α sheet has been proposed as the toxic constituent of amyloid precursor proteins,[9] but this idea is not widely accepted at present. 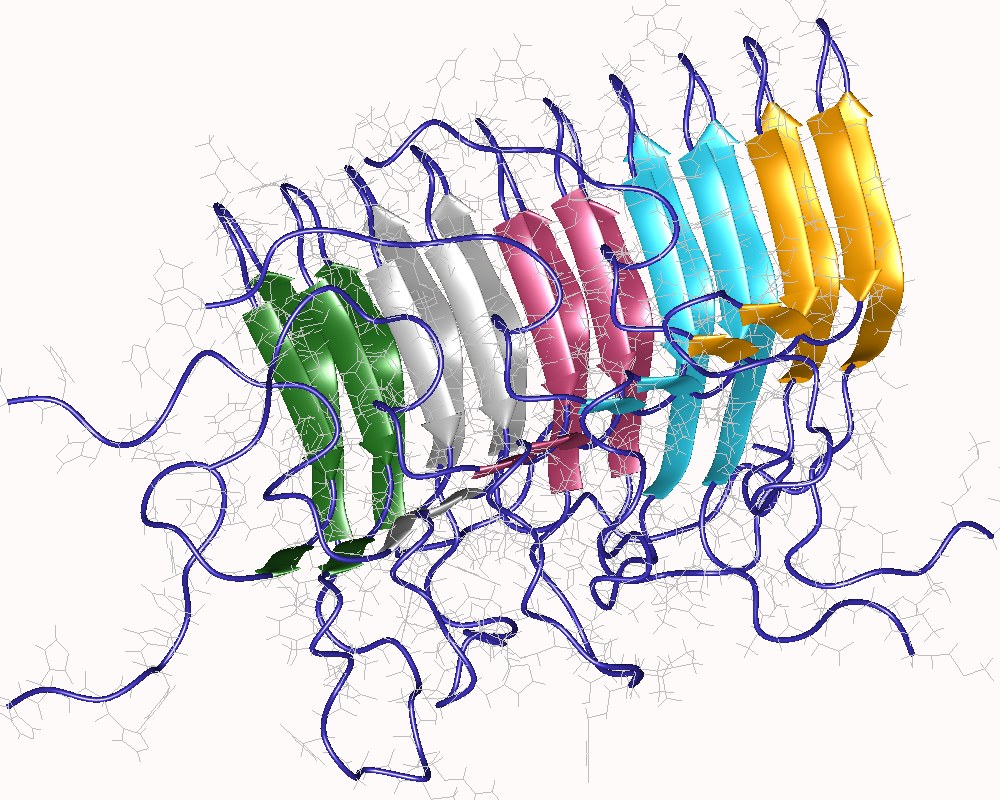 Amyloid of HET-s(218-289) prion pentamer, Podospora anserina (PDB: 2rnm) Definition The name amyloid comes from the early mistaken identification by Rudolf Virchow of the substance as starch (amylum in Latin, from Ancient Greek: ἄμυλον, romanized: amylon), based on crude iodine-staining techniques. For a period, the scientific community debated whether or not amyloid deposits are fatty deposits or carbohydrate deposits until it was finally found (in 1859) that they are, in fact, deposits of albumoid proteinaceous material.[10] The classical, histopathological definition of amyloid is an extracellular, proteinaceous fibrillar deposit exhibiting β-sheet secondary structure and identified by apple-green birefringence when stained with congo red under polarized light. These deposits often recruit various sugars and other components such as serum amyloid P component, resulting in complex, and sometimes inhomogeneous structures.[11] Recently this definition has come into question as some classic, amyloid species have been observed in distinctly intracellular locations.[12] A more recent, biophysical definition is broader, including any polypeptide that polymerizes to form a cross-β structure, in vivo or in vitro, inside or outside cells. Microbiologists, biochemists, biophysicists, chemists and physicists have largely adopted this definition,[13][14] leading to some conflict in the biological community over an issue of language. |
アミロイドはタンパク質の凝集体で、通常直径7~13nmの線維状形
態、βシート二次構造(クロスβと呼ばれる)、コンゴーレッドなどの特定の色素による染色性を特徴とする[1]
。このようなタンパク質のミスフォールディングや沈着は、組織や臓器の健全な機能を破壊する。 このようなアミロイドは、アミロイドーシスとして知られる50を超える[2][3]ヒトの疾患と関連しており(必ずしもその原因とは限らない)、神経変性 疾患の一部にも関与している可能性がある[2][4]。また、家族性のみのものもある。医療行為に起因するものもある。プリオンはアミロイドの感染型であ り、他の非感染型を変換する鋳型として働くことができる[5]。アミロイドは通常の生物学的機能を持つこともある。例えば、いくつかの属の細菌における フィンブリア形成、真菌におけるエピジェネティック形質の伝達、ヒトにおける色素沈着やホルモン放出などである[6]。 アミロイドは多くの異なるタンパク質から生じることが知られている[2][7]。これらのポリペプチド鎖は一般的にβシート構造を形成し、長い繊維状に凝 集するが、同一のポリペプチドが複数の異なるアミロイド構造に折り畳まれることもある[8]。 αシートと呼ばれる珍しい二次構造がアミロイド前駆体タンパク質の毒性成分として提唱されているが[9]、この考えは現在のところあまり受け入れられてい ない。 HET-s(218-289)プリオン5量体のアミロイド、Podospora anserina (PDB: 2rnm) 定義 アミロイドという名称は、ルドルフ・ヴィルヒョー(Rudolf Virchow)が、粗雑なヨード染色技術に基づいて、この物質をデンプン(ラテン語ではamylum、古代ギリシャ語ではἄμυλον, ローマ字ではamylon)と誤って同定したことに由来する。一時期、科学界では、アミロイド沈着物が脂肪沈着物なのか、炭水化物沈着物なのかが議論され たが、最終的に(1859年に)、アミロイド沈着物は実際にはアルブモイド蛋白質の沈着物であることが判明した[10]。 アミロイドの古典的な病理組織学的定義は、βシート二次構造を示す細胞外のタンパク質性線維性沈着物であり、偏光下でコンゴーレッドで染色するとアップル グリーンの複屈折によって同定される。これらの沈着物は、しばしば様々な糖や血清アミロイドP成分などの他の成分を取り込み、複雑で、時には不均一な構造 をもたらす[11]。最近、いくつかの古典的なアミロイド種が細胞内のはっきりとした場所で観察されたことから、この定義が疑問視されている[12]。 より最近の、生物物理学的な定義はより広範で、in vivoでもin vitroでも、細胞内でも細胞外でも、重合してクロスβ構造を形成するあらゆるポリペプチドを含む。微生物学者、生化学者、生物物理学者、化学者、物理 学者はこの定義をほぼ採用しており[13][14]、生物学界では言葉の問題をめぐっていくつかの対立が生じている。 |
| Proteins forming amyloids in
diseases To date, 37 human proteins have been found to form amyloid in pathology and be associated with well-defined diseases.[2] The International Society of Amyloidosis classifies amyloid fibrils and their associated diseases based upon associated proteins (for example ATTR is the group of diseases and associated fibrils formed by TTR).[3] A table is included below. (以下省略)https://en.wikipedia.org/wiki/Amyloid |
疾患においてアミロイドを形成するタンパク質 現在までに、37のヒトタンパク質が病理学的にアミロイドを形成し、明確に定義された疾患と関連していることが判明している[2]。国際アミロイドーシス 学会は、アミロイド線維とその関連疾患を関連タンパク質に基づいて分類している(例えば、ATTRはTTRによって形成される疾患群と関連線維)[3]。 (以下省略)https://en.wikipedia.org/wiki/Amyloid |
| Non-disease and functional
amyloids (以下省略)https://en.wikipedia.org/wiki/Amyloid |
非疾患と機能的アミロイド(以下省略)https://en.wikipedia.org/wiki/Amyloid |
Structure Structure of a fibril, consisting of one single protofilament, of the amyloid β peptide viewed down the long axis of the fibril (PDB: 2mlq)[44] Amyloids are formed of long unbranched fibers that are characterized by an extended β-sheet secondary structure in which individual β strands (β-strands) (coloured arrows in the adjacent figure) are arranged in an orientation perpendicular to the long axis of the fiber. Such a structure is known as cross-β structure. Each individual fiber may be 7–13 nanometres in width and a few micrometres in length.[6][2] The main hallmarks recognised by different disciplines to classify protein aggregates as amyloid is the presence of a fibrillar morphology with the expected diameter, detected using transmission electron microscopy (TEM) or atomic force microscopy (AFM), the presence of a cross-β secondary structure, determined with circular dichroism, FTIR, solid-state nuclear magnetic resonance (ssNMR), X-ray crystallography, or X-ray fiber diffraction (often considered the "gold-standard" test to see whether a structure contains cross-β fibres), and an ability to stain with specific dyes, such as Congo red, thioflavin T or thioflavin S.[2] The term "cross-β" was based on the observation of two sets of diffraction lines, one longitudinal and one transverse, that form a characteristic "cross" pattern.[45] There are two characteristic scattering diffraction signals produced at 4.7 and 10 Å (0.47 nm and 1.0 nm), corresponding to the interstrand and stacking distances in β sheets.[1] The "stacks" of β sheet are short and traverse the breadth of the amyloid fibril; the length of the amyloid fibril is built by aligned β-strands. The cross-β pattern is considered a diagnostic hallmark of amyloid structure.[6] Amyloid fibrils are generally composed of 1–8 protofilaments (one protofilament also corresponding to a fibril is shown in the figure), each 2–7 nm in diameter, that interact laterally as flat ribbons that maintain the height of 2–7 nm (that of a single protofilament) and are up to 30 nm wide; more often protofilaments twist around each other to form the typically 7–13 nm wide fibrils.[2] Each protofilament possesses the typical cross-β structure and may be formed by 1–6 β-sheets (six are shown in the figure) stacked on each other. Each individual protein molecule can contribute one to several β-strands in each protofilament and the strands can be arranged in antiparallel β-sheets, but more often in parallel β-sheets. Only a fraction of the polypeptide chain is in a β-strand conformation in the fibrils, the remainder forms structured or unstructured loops or tails. For a long time our knowledge of the atomic-level structure of amyloid fibrils was limited by the fact that they are unsuitable for the most traditional methods for studying protein structures. Recent years have seen progress in experimental methods, including solid-state NMR spectroscopy and Cryo-Electron Microscopy. Combined, these methods have provided 3D atomic structures of amyloid fibrils formed by amyloid β peptides, α-synuclein, tau, and the FUS protein, associated with various neurodegenerative diseases.[46][47] X-ray diffraction studies of microcrystals revealed atomistic details of core region of amyloid, although only for simplified peptides having a length remarkably shorter than that of peptides or proteins involved in disease.[48][49] The crystallographic structures show that short stretches from amyloid-prone regions of amyloidogenic proteins run perpendicular to the filament axis, consistent with the "cross-β" feature of amyloid structure. They also reveal a number of characteristics of amyloid structures – neighboring β-sheets are tightly packed together via an interface devoid of water (therefore referred to as dry interface), with the opposing β-strands slightly offset from each other such that their side-chains interdigitate. This compact dehydrated interface created was termed a steric-zipper interface.[6] There are eight theoretical classes of steric-zipper interfaces, dictated by the directionality of the β-sheets (parallel and anti-parallel) and symmetry between adjacent β-sheets. A limitation of X-ray crystallography for solving amyloid structure is represented by the need to form microcrystals, which can be achieved only with peptides shorter than those associated with disease. Although bona fide amyloid structures always are based on intermolecular β-sheets, different types of "higher order" tertiary folds have been observed or proposed. The β-sheets may form a β-sandwich, or a β-solenoid which may be either β-helix or β-roll. Native-like amyloid fibrils in which native β-sheet containing proteins maintain their native-like structure in the fibrils have also been proposed.[50] There are few developed ideas on how the complex backbone topologies of disulfide-constrained proteins, which are prone to form amyloid fibrils (such as insulin and lysozyme), adopt the amyloid β-sheet motif. The presence of multiple constraints significantly reduces the accessible conformational space, making computational simulations of amyloid structures more feasible. [51] One complicating factor in studies of amyloidogenic polypeptides is that identical polypeptides can fold into multiple distinct amyloid conformations.[6] This phenomenon is typically described as amyloid polymorphism.[8][52] [53] It has notable biological consequences given that it is thought to explain the prion strain phenomenon. |
構造 1本のプロトフィラメントからなるアミロイドβペプチドのフィブリルの長軸方向の構造(PDB: 2mlq)[44]。 アミロイドは分枝していない長い線維で形成され、個々のβストランド(β鎖)(隣の図では色のついた矢印)が線維の長軸に垂直な方向に配列した、拡張した βシート二次構造が特徴である。このような構造はクロスβ構造として知られている。個々の繊維の幅は7~13ナノメートル、長さは数マイクロメートルであ る。 [6][2]タンパク質の凝集体をアミロイドとして分類するために様々な分野で認識されている主な特徴は、透過型電子顕微鏡(TEM)または原子間力顕微 鏡(AFM)を用いて検出される、予想される直径を持つ線維状の形態の存在、クロスβ二次構造の存在である、 円偏光二色性、FTIR、固体核磁気共鳴(ssNMR)、X線結晶学、X線繊維回折(構造がクロスβ繊維を含むかどうかを見るための「ゴールドスタンダー ド」テストと考えられている)、コンゴーレッド、チオフラビンT、チオフラビンSなどの特定の色素で染色できることである。 [2] クロスβ」という用語は、特徴的な「クロス」パターンを形成する縦方向と横方向の2組の回折線の観察に基づいている[45]。 7Åと10Å(0.47 nmと1.0 nm)には、βシートの鎖間距離と積層距離に対応する2つの特徴的な散乱回折シグナルが生じる[1]。βシートの「積層」は短く、アミロイド線維の幅を横 切る。クロスβパターンは、アミロイド構造の診断上の特徴であると考えられている[6]。 アミロイド線維は一般的に、直径2-7 nmの1-8本のプロトフィラメント(図には線維に相当するプロトフィラメントも1本示されている)から構成され、それらは2-7 nmの高さ(1本のプロトフィラメントの高さ)を維持し、最大30 nmの幅を持つ平坦なリボンとして横方向に相互作用する。 [2] 各プロトフィラメントは典型的なクロスβ構造を持ち、1-6枚のβシート(図では6枚が示されている)が重なり合って形成されている。個々のタンパク質分 子は、各プロトフィラメントにおいて1本から数本のβ-ストランドに寄与し、ストランドは反平行β-シートに配列することができるが、より多くの場合、平 行β-シートに配列する。フィブリル中でβ-ストランド構造をとるポリペプチド鎖はごく一部であり、残りは構造化された、あるいは構造化されていないルー プやテールを形成している。 長い間、アミロイド線維の原子レベルの構造に関する知識は、タンパク質の構造を研究する最も伝統的な方法には適さないという事実によって制限されていた。 近年、固体NMR分光法や低温電子顕微鏡法などの実験的手法が進歩した。これらの方法を組み合わせることで、様々な神経変性疾患に関連するアミロイドβペ プチド、αシヌクレイン、タウ、FUSタンパク質によって形成されるアミロイド線維の3次元原子構造が得られている[46][47]。 微小結晶のX線回折研究により、アミロイドのコア領域の原子論的な詳細が明らかになったが、それは疾患に関与するペプチドやタンパク質よりも著しく短い長 さの単純化されたペプチドについてのみであった[48][49]。結晶構造は、アミロイド生成タンパク質のアミロイドになりやすい領域からの短いストレッ チがフィラメント軸に対して垂直に走っていることを示しており、アミロイド構造の「クロスβ」特徴と一致している。また、アミロイド構造の多くの特徴も明 らかになった。隣り合うβシートは、水のない界面(したがって乾燥界面と呼ばれる)を介してしっかりと密着し、対向するβストランドは互いにわずかにずれ ており、側鎖が相互接続する。このコンパクトな脱水界面は、立体-ジッパー界面と呼ばれる[6]。立体-ジッパー界面には、βシートの方向性(平行と反平 行)と隣接するβシート間の対称性によって決まる8つの理論的クラスがある。アミロイドの構造を解明するためのX線結晶構造解析の限界は、微結晶を形成す る必要性に代表されるが、これは疾患に関連するペプチドよりも短いペプチドでしか達成できない。 真正のアミロイド構造は常に分子間βシートに基づいているが、異なるタイプの 「高次 」3次フォールドが観察されたり提案されている。βシートはβサンドイッチを形成することもあれば、β-ヘリックスまたはβ-ロールを形成するβ-ソレノ イドを形成することもある。アミロイド線維を形成しやすいジスルフィド拘束タンパク質(インスリンやリゾチームなど)の複雑な骨格トポロジーが、どのよう にしてアミロイドβシートモチーフを採用するのかについて、発展したアイデアはほとんどない。複数の拘束が存在することで、アクセス可能なコンフォメー ション空間が著しく減少し、アミロイド構造の計算シミュレーションがより実行しやすくなる。[51] アミロイド生成ポリペプチドの研究を複雑にしている要因の一つは、同一のポリペプチドが複数の異なるアミロイドコンフォメーションに折り畳まれる可能性が あることである。 |
| Amino acid sequence and amyloid
formation In general, amyloid polymerization (aggregation or non-covalent polymerization) is sequence-sensitive, that is mutations in the sequence can induce or prevent self-assembly.[61][62] For example, humans produce amylin, an amyloidogenic peptide associated with type II diabetes, but in rats and mice prolines are substituted in critical locations and amyloidogenesis does not occur.[citation needed] Studies comparing synthetic to recombinant β amyloid peptide in assays measuring rate of fibrillation, fibril homogeneity, and cellular toxicity showed that recombinant β amyloid peptide has a faster fibrillation rate and greater toxicity than synthetic β amyloid peptide.[63] There are multiple classes of amyloid-forming polypeptide sequences.[8][52][53] Glutamine-rich polypeptides are important in the amyloidogenesis of Yeast and mammalian prions, as well as trinucleotide repeat disorders including Huntington's disease. When glutamine-rich polypeptides are in a β-sheet conformation, glutamines can brace the structure by forming inter-strand hydrogen bonding between its amide carbonyls and nitrogens of both the backbone and side chains. The onset age for Huntington's disease shows an inverse correlation with the length of the polyglutamine sequence, with analogous findings in a C. elegans model system with engineered polyglutamine peptides.[64] Other polypeptides and proteins such as amylin and the β amyloid peptide do not have a simple consensus sequence and are thought to aggregate through the sequence segments enriched with hydrophobic residues, or residues with high propensity to form β-sheet structure.[61] Among the hydrophobic residues, aromatic amino-acids are found to have the highest amyloidogenic propensity.[65][66] Cross-polymerization (fibrils of one polypeptide sequence causing other fibrils of another sequence to form) is observed in vitro and possibly in vivo. This phenomenon is important, since it would explain interspecies prion propagation and differential rates of prion propagation, as well as a statistical link between Alzheimer's and type 2 diabetes.[67] In general, the more similar the peptide sequence the more efficient cross-polymerization is, though entirely dissimilar sequences can cross-polymerize and highly similar sequences can even be "blockers" that prevent polymerization.[citation needed] Amyloid toxicity The reasons why amyloid cause diseases are unclear. In some cases, the deposits physically disrupt tissue architecture, suggesting disruption of function by some bulk process. An emerging consensus implicates prefibrillar intermediates, rather than mature amyloid fibers, in causing cell death, particularly in neurodegenerative diseases.[17][68] The fibrils are, however, far from innocuous, as they keep the protein homeostasis network engaged, release oligomers, cause the formation of toxic oligomers via secondary nucleation, grow indefinitely spreading from district to district[2] and, in some cases, may be toxic themselves.[69] Calcium dysregulation has been observed to occur early in cells exposed to protein oligomers. These small aggregates can form ion channels through lipid bilayer membranes and activate NMDA and AMPA receptors. Channel formation has been hypothesized to account for calcium dysregulation and mitochondrial dysfunction by allowing indiscriminate leakage of ions across cell membranes.[70] Studies have shown that amyloid deposition is associated with mitochondrial dysfunction and a resulting generation of reactive oxygen species (ROS), which can initiate a signalling pathway leading to apoptosis.[71] There are reports that indicate amyloid polymers (such as those of huntingtin, associated with Huntington's disease) can induce the polymerization of essential amyloidogenic proteins, which should be deleterious to cells. Also, interaction partners of these essential proteins can also be sequestered.[72] All these mechanisms of toxicity are likely to play a role. In fact, the aggregation of a protein generates a variety of aggregates, all of which are likely to be toxic to some degree. A wide variety of biochemical, physiological and cytological perturbations has been identified following the exposure of cells and animals to such species, independently of their identity. The oligomers have also been reported to interact with a variety of molecular targets. Hence, it is unlikely that there is a unique mechanism of toxicity or a unique cascade of cellular events. The misfolded nature of protein aggregates causes a multitude of aberrant interactions with a multitude of cellular components, including membranes, protein receptors, soluble proteins, RNAs, small metabolites, etc. Histological staining In the clinical setting, amyloid diseases are typically identified by a change in the spectroscopic properties of planar aromatic dyes such as thioflavin T, congo red or NIAD-4.[73] In general, this is attributed to the environmental change, as these dyes intercalate between β-strands to confine their structure.[74] Congo Red positivity remains the gold standard for diagnosis of amyloidosis. In general, binding of Congo Red to amyloid plaques produces a typical apple-green birefringence when viewed under cross-polarized light. Recently, significant enhancement of fluorescence quantum yield of NIAD-4 was exploited to super-resolution fluorescence imaging of amyloid fibrils[75] and oligomers.[76] To avoid nonspecific staining, other histology stains, such as the hematoxylin and eosin stain, are used to quench the dyes' activity in other places such as the nucleus, where the dye might bind. Modern antibody technology and immunohistochemistry has made specific staining easier, but often this can cause trouble because epitopes can be concealed in the amyloid fold; in general, an amyloid protein structure is a different conformation from the one that the antibody recognizes. |
アミノ酸配列とアミロイド形成 一般に、アミロイド重合(凝集または非共有結合重合)は塩基配列に敏感であり、すなわち塩基配列の変異が自己集合を誘導したり阻害したりする[61] [62]。例えば、ヒトはII型糖尿病に関連するアミロイド生成ペプチドであるアミリンを生成するが、ラットやマウスではプロリンが重要な位置で置換され ており、アミロイド生成は起こらない。 [引用が必要]線維化速度、線維の均一性、細胞毒性を測定するアッセイで合成βアミロイドペプチドと組換えβアミロイドペプチドを比較した研究では、組換 えβアミロイドペプチドは合成βアミロイドペプチドよりも線維化速度が速く、毒性も大きいことが示された[63]。 アミロイド形成ポリペプチド配列には複数のクラスがある[8][52][53]。グルタミンに富んだポリペプチドは、酵母や哺乳類のプリオン、およびハン チントン病を含む3塩基反復障害のアミロイド形成において重要である。グルタミンを多く含むポリペプチドがβシートコンフォメーションにあるとき、グルタ ミンはそのアミドカルボニルと骨格および側鎖の両方のニトロゲンとの間で鎖間水素結合を形成することにより、構造を支えることができる。ハンチントン病の 発症年齢は、ポリグルタミン配列の長さと逆相関を示し、ポリグルタミンペプチドを操作した線虫モデル系でも同様の所見が得られている[64]。 アミリンやβアミロイドペプチドのような他のポリペプチドやタンパク質は単純なコンセンサス配列を持たず、疎水性残基やβシート構造を形成する傾向の高い 残基に富む配列セグメントを介して凝集すると考えられている[61]。疎水性残基の中で、芳香族アミノ酸が最もアミロイド形成傾向が高いことが分かってい る[65][66]。 交差重合(あるポリペプチド配列のフィブリルが別の配列のフィブリルを形成させる)はin vitroで観察され、おそらくin vivoでも観察される。一般に、ペプチド配列が類似しているほど交差重合は効率的であるが、全く異なる配列が交差重合することもあり、非常に類似した配 列が重合を妨げる「ブロッカー」になることさえある[67]。 アミロイド毒性 アミロイドが病気を引き起こす理由は不明である。場合によっては、沈着物が物理的に組織構造を破壊し、何らかのバルクプロセスによる機能破壊を示唆してい る。特に神経変性疾患では、成熟したアミロイド線維よりもむしろ、前線維の中間体が細胞死を引き起こすことに関与するというのが、新たなコンセンサスであ る[17][68]。しかしながら、線維は、タンパク質のホメオスタシスネットワークを維持し、オリゴマーを放出し、二次核形成を介して有毒なオリゴマー の形成を引き起こし、地区から地区へと広がりながら無限に成長し[2]、場合によってはそれ自体が有毒であることもあるため、無害とは言い難い[69]。 カルシウム調節障害は、タンパク質オリゴマーに曝された細胞で早期に起こることが観察されている。これらの小さな凝集体は、脂質二重膜を通してイオンチャ ネルを形成し、NMDA受容体やAMPA受容体を活性化する。チャネルの形成は、細胞膜を介したイオンの無差別的な漏出を可能にすることにより、カルシウ ム調節異常とミトコンドリア機能不全の原因であるという仮説が立てられている。 [アミロイドポリマー(ハンチントン病に関連するハンチンチンなど)は、アミロイド形成に必須なタンパク質の重合を誘導し、細胞にとって有害であることを 示す報告がある。また、これらの必須タンパク質の相互作用パートナーも隔離される可能性がある。 このような毒性のメカニズムがすべて関与している可能性が高い。実際、タンパク質の凝集は様々な凝集体を生成し、その全てがある程度の毒性を持つ可能性が 高い。細胞や動物がこのような凝集体にさらされると、その正体とは無関係に、生化学的、生理学的、細胞学的に様々な障害が起こることが確認されている。ま た、オリゴマーは様々な分子標的と相互作用することが報告されている。従って、毒性の特異的なメカニズムや細胞内事象の特異的なカスケードが存在するとは 考えにくい。タンパク質凝集体のミスフォールディングの性質は、膜、タンパク質レセプター、可溶性タンパク質、RNA、低分子代謝産物など、多数の細胞成 分と多数の異常な相互作用を引き起こす。 組織学的染色 臨床の場では、アミロイド疾患は通常、チオフラビンT、コンゴーレッド、NIAD-4などの平面芳香族色素の分光学的特性の変化によって同定される [73]。一般に、これは、これらの色素がβストランドの間にインターカレートしてその構造を閉じ込めることによる環境変化に起因する[74]。 コンゴーレッド陽性は、アミロイドーシス診断のゴールドスタンダードであり続けている。一般に、コンゴーレッドがアミロイド斑に結合すると、交差偏光下で 見たときに典型的なアップルグリーンの複屈折が生じる。最近、NIAD-4の蛍光量子収率の大幅な向上が、アミロイド線維[75]やオリゴマーの超解像蛍 光イメージングに利用された[76]。非特異的な染色を避けるために、ヘマトキシリン・エオジン染色などの他の組織染色が、色素が結合する可能性のある核 などの他の場所での色素の活性を消光するために使用される。現代の抗体技術と免疫組織化学は、特異的染色を容易にしたが、エピトープがアミロイドフォール ドの中に隠されていることがあるため、しばしば問題を引き起こすことがある。 |
| APOE(Apolipoprotein E):アポリポ蛋白E Apolipoprotein E (Apo-E) is a protein involved in the metabolism of fats in the body of mammals. A subtype is implicated in Alzheimer's disease and cardiovascular diseases.[5] It is encoded in humans by the gene APOE. Apo-E belongs to a family of fat-binding proteins called apolipoproteins. In the circulation, it is present as part of several classes of lipoprotein particles, including chylomicron remnants, VLDL, IDL, and some HDL.[6] APOE interacts significantly with the low-density lipoprotein receptor (LDLR), which is essential for the normal processing (catabolism) of triglyceride-rich lipoproteins.[7] In peripheral tissues, APOE is primarily produced by the liver and macrophages, and mediates cholesterol metabolism. In the central nervous system, Apo-E is mainly produced by astrocytes and transports cholesterol to neurons[8] via APOE receptors, which are members of the low density lipoprotein receptor gene family.[9] Apo-E is the principal cholesterol carrier in the brain.[10] APOE qualifies as a checkpoint inhibitor of the classical complement pathway by complex formation with activated C1q.[11] Evolution Apolipoproteins are not unique to mammals. Many terrestrial and marine vertebrates have versions of them.[12] It is believed that APOE arose via gene duplications of APOC1 before the fish–tetrapod split ca. 400 million years ago. Proteins similar in function have been found in choanoflagellates, suggesting that they are a very old class of proteins predating the dawn of all living animals.[13] The three major human alleles (E4, E3, E2) arose after the primate–human split around 7.5 million years ago. These alleles are the by-product of non-synonymous mutations which led to changes in functionality. The first allele to emerge was E4. After the primate–human split, there were four amino acid changes in the human lineage, three of which had no effect on protein function (V174L, A18T, A135V). The fourth substitution (T61R) traded a threonine for an arginine altering the protein's functionality. This substitution occurred somewhere in the 6 million year gap between the primate–human split and the Denisovan–human split, since exactly the same substitutions were found in Denisovan APOE.[14] About 220,000 years ago, a cysteine to arginine substitution took place at amino acid 112 (Cys112Arg) of the APOE4 gene, and this resulted in the E3 allele. Finally, 80,000 years ago, another arginine to cysteine substitution at amino acid 158 (Arg158Cys) of the APOE3 gene created the E2 allele.[15][13] Structure Gene The gene, APOE, is mapped to chromosome 19 in a cluster with the apolipoprotein C1 (APOC1) gene and the apolipoprotein C2 (APOC2) gene. The APOE gene consists of four exons and three introns, totaling 3597 base pairs. APOE is transcriptionally activated by the liver X receptor (an important regulator of cholesterol, fatty acid, and glucose homeostasis) and peroxisome proliferator-activated receptor γ, nuclear receptors that form heterodimers with retinoid X receptors.[16] In melanocytic cells APOE gene expression may be regulated by MITF.[17] Protein APOE is 299 amino acids long and contains multiple amphipathic α-helices. According to crystallography studies, a hinge region connects the N- and C-terminal regions of the protein. The N-terminal region (residues 1–167) forms an anti-parallel four-helix bundle such that the non-polar sides face inside the protein. Meanwhile, the C-terminal domain (residues 206–299) contains three α-helices which form a large exposed hydrophobic surface and interact with those in the N-terminal helix bundle domain through hydrogen bonds and salt-bridges. The C-terminal region also contains a low density lipoprotein receptor (LDLR)-binding site.[18] |
アポリポ蛋白質E(Apo-E)は、哺乳類の体内で脂肪の代謝に関与する蛋白質である。ヒトではAPOE遺伝子によってコードされている。 Apo-Eは、アポリポタンパク質と呼ばれる脂肪結合タンパク質のファミリーに属する。循環中では、カイロミクロン残骸、VLDL、IDL、および一部の HDLを含むいくつかのクラスのリポ蛋白粒子の一部として存在する[6]。APOEは、トリグリセリドに富むリポ蛋白の正常な処理(異化)に必須である低 比重リポ蛋白受容体(LDLR)と有意に相互作用する[7]。末梢組織では、APOEは主に肝臓とマクロファージによって産生され、コレステロール代謝を 媒介する。中枢神経系では、Apo-Eは主にアストロサイトによって産生され、低密度リポ蛋白質受容体遺伝子ファミリーのメンバーであるAPOE受容体を 介して、コレステロールを神経細胞に輸送する[8]。 進化 アポリポ蛋白質は哺乳類特有のものではない。APOEは、約4億年前に魚類と四肢動物が分裂する前に、APOC1の遺伝子重複を経て誕生したと考えられて いる[12]。4億年前である。同様の機能を持つタンパク質が襟鞭毛虫で見つかっており、このことは、APOEが、すべての生物の夜明けよりも前に存在し た、非常に古いタンパク質の一種であることを示唆している[13]。 ヒトの3つの主要な対立遺伝子(E4、E3、E2)は、約750万年前の霊長類とヒトの分裂後に生じた。これらの対立遺伝子は、機能性の変化をもたらした 非同義変異の副産物である。最初に出現した対立遺伝子はE4である。霊長類とヒトの分裂後、ヒトの系統では4つのアミノ酸の変化があったが、そのうちの3 つはタンパク質の機能に影響を与えなかった(V174L、A18T、A135V)。4番目の置換(T61R)はスレオニンをアルギニンと交換し、タンパク 質の機能を変えた。この置換は、霊長類-ヒトの分裂とデニソワ人-ヒトの分裂の間の600万年のギャップのどこかで起こった。 約22万年前、APOE4遺伝子のアミノ酸112(Cys112Arg)でシステインからアルギニンへの置換が起こり、E3対立遺伝子が生じた。最後に、 8万年前に、APOE3遺伝子のアミノ酸158(Arg158Cys)でアルギニンからシステインへの置換が起こり、E2対立遺伝子が生じた[15] [13]。 構造 遺伝子 APOE遺伝子は、アポリポタンパク質C1(APOC1)遺伝子およびアポリポタンパク質C2(APOC2)遺伝子と一緒に19番染色体にマッピングされ ている。APOE遺伝子は4つのエクソンと3つのイントロンからなり、合計3597塩基対である。APOEは、レチノイドX受容体とヘテロ二量体を形成す る核内受容体である肝X受容体(コレステロール、脂肪酸、グルコースの恒常性の重要な調節因子)およびペルオキシソーム増殖因子活性化受容体γによって転 写活性化される[16]。 タンパク質 APOEは299アミノ酸長で、複数の両親媒性α-ヘリックスを含む。結晶学的研究によると、ヒンジ領域がタンパク質のN-末端領域とC-末端領域をつな いでいる。N末端領域(残基1-167)は、非極性側がタンパク質の内側を向くように、反平行4らせんの束を形成している。一方、C末端領域(残基206 -299)には3つのα-ヘリックスがあり、大きな疎水性表面を露出させ、水素結合や塩架橋を介してN末端ヘリックスバンドル領域のα-ヘリックスと相互 作用する。C末端領域には低密度リポタンパク質受容体(LDLR)結合部位もある[18]。 |
| Much
remains to be learned about the APOE isoforms, including the
interaction of other protective genes.[58] Indeed, the apolipoprotein
ε4 isoform is more protective against cognitive decline than other
isoforms in some cases,[58] so caution is advised before making
determinant statements about the influence of APOE polymorphisms on
cognition, development of Alzheimer's disease, cardiovascular disease,
telomere shortening, etc. Many of the studies cited that purport these
adverse outcomes are from single studies that have not been replicated
and the research is based on unchecked assumptions about this isoform.
As of 2007, there was no evidence that APOE polymorphisms influence
cognition in younger age groups (other than possible increased episodic
memory ability and neural efficiency in younger APOE4 age groups), nor
that the APOE4 isoform places individuals at increased risk for any
infectious disease.[59] However, the association between the APOE4 allele and Alzheimer's disease has been shown to be weaker in minority groups differently compared to their Caucasian counterparts.[9] Hispanics/Latinos and African Americans who were homozygous for the APOE4 allele had 2.2 and 5.7 times the odds, respectively of developing Alzheimer's disease.[60][9] The APOE4 allele has an even stronger effect in East Asian populations, with Japanese populations have 33 times the odds compared to other populations.[61] Caucasians who were homozygous for the allele had 12.5 times the odds.[60][9] Function As a component of the lipoprotein lipid transport system, APOE facilitates the transport of lipids, fat-soluble vitamins, and cholesterol via the blood. It interacts with the LDL receptor to facilitate endocytosis of VLDL remnants. It is synthesized principally in the liver, but has also been found in other tissues such as the brain, kidneys, and spleen.[21] APOE synthesized in the liver associates with HDL which can then distribute it to newly formed VLDL or chylomicron particles to facilitate their eventual uptake by the liver. In the nervous system, non-neuronal cell types, most notably astroglia and microglia, are the primary producers of APOE, while neurons preferentially express the receptors for APOE.[62] There are seven currently identified mammalian receptors for APOE which belong to the evolutionarily conserved LDLR family.[63] APOE was initially recognized for its importance in lipoprotein metabolism and cardiovascular disease. Defects in APOE result in familial dysbetalipoproteinemia aka type III hyperlipoproteinemia (HLP III), in which increased plasma cholesterol and triglycerides are the consequence of impaired clearance of chylomicron, VLDL and LDL.[64][7] More recently, it has been studied for its role in several biological processes not directly related to lipoprotein transport, including Alzheimer's disease (AD), immunoregulation, and cognition.[5] Though the exact mechanisms remain to be elucidated, isoform 4 of APOE, encoded by an APOE allele, has been associated with increased calcium ion levels and apoptosis following mechanical injury.[65] In the field of immune regulation, a growing number of studies point to APOE's interaction with many immunological processes, including suppressing T cell proliferation, macrophage functioning regulation, lipid antigen presentation facilitation (by CD1)[66] to natural killer T cell as well as modulation of inflammation and oxidation.[67] APOE is produced by macrophages and APOE secretion has been shown to be restricted to classical monocytes in PBMC, and the secretion of APOE by monocytes is down regulated by inflammatory cytokines and upregulated by TGF-beta.[68] Clinical significance Alzheimer's disease As of 2012, the E4 variant was the largest known genetic risk factor for late-onset sporadic Alzheimer's disease (AD) in a variety of ethnic groups.[69] However, the E4 variant does not correlate with risk in every population. Nigerian people have the highest observed frequency of the APOE4 allele in world populations,[70] but AD is rare among them.[70][71] This may be due to their low cholesterol levels.[70][71][72][73] Caucasian and Japanese carriers of two E4 alleles have between 10 and 30 times the risk of developing AD by 75 years of age, as compared to those not carrying any E4 alleles. This may be caused by an interaction with amyloid.[74] Alzheimer's disease is characterized by build-ups of aggregates of the peptide beta-amyloid. Apolipoprotein E enhances proteolytic break-down of this peptide, both within and between cells. The isoform APOE-ε4 is not as effective as the others at promoting these reactions, resulting in increased vulnerability to AD in individuals with that gene variation.[75] Recently, the amyloid hypothesis of Alzheimer's disease has been questioned, and an article in Science claimed that "Just as removing smoke does not extinguish a fire, reducing amyloid plaques may not affect the course of Alzheimer's disease."[76] The role that the E4 variant carries can still be fully explained even in the absence of a valid amyloid hypothesis given the fact that reelin signaling emerges to be one of the key processes involved in Alzheimer's disease[77] and the E4 variant is shown to interact with ApoER2, one of the neuronal reelin receptors, thereby obstructing reelin signaling.[77] Although 40–65% of AD patients have at least one copy of the ε4 allele, APOE4 is not a determinant of the disease. At least one-third of patients with AD are APOE4 negative and some APOE4 homozygotes never develop the disease. Yet those with two ε4 alleles have up to 20 times the risk of developing AD.[78] There is also evidence that the APOE2 allele may serve a protective role in AD.[79] Thus, the genotype most at risk for Alzheimer's disease and at an earlier age is APOE4,4. Using genotype APOE3,3 as a benchmark (with the persons who have this genotype regarded as having a risk level of 1.0) and for white populations only, individuals with genotype APOE4,4 have an odds ratio of 14.9 of developing Alzheimer's disease. Individuals with the APOE3,4 genotype face an odds ratio of 3.2, and people with a copy of the 2 allele and the 4 allele (APOE2,4), have an odds ratio of 2.6. Persons with one copy each of the 2 allele and the 3 allele (APOE2,3) have an odds ratio of 0.6. Persons with two copies of the 2 allele (APOE2,2) also have an odds ratio of 0.6.[80] 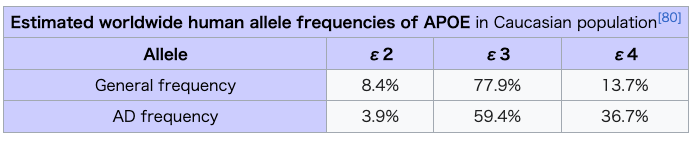 While ApoE4 has been found to greatly increase the odds that an individual will develop Alzheimer's, a 2002 study concluded, that in persons with any combination of APOE alleles, high serum total cholesterol and high blood pressure in mid-life are independent risk factors which together can nearly triple the risk that the individual will later develop AD.[73] Projecting from their data, some researchers have suggested that lowering serum cholesterol levels may reduce a person's risk for Alzheimer's disease, even if they have two ApoE4 alleles, thus reducing the risk from nine or ten times the odds of getting AD down to just two times the odds.[73] Women are more likely to develop AD than men across most ages and APOE genotypes. Premorbid women with the ε4 allele have significantly more neurological dysfunction than men.[81] APOE-ε4 increases the risk not only for AD but also for dementia in pure alpha-synucleinopathies.[82] The influence of APOE-ε4 on hippocampal atrophy was suggested to be more predominant early in the course of AD at milder stages prior to more widespread neurodegeneration.[83] Atherosclerosis Knockout mice that lack the apolipoprotein-E gene (APOE−/−) develop extreme hypercholesterolemia when fed a high-fat diet.[84] Malaria APOE−/− knockout mice show marked attenuation of cerebral malaria and increased survival, as well as decreased sequestration of parasites and T cells within the brain, likely due to protection of the blood–brain barrier.[85] Human studies have shown that the APOE2 polymorphism correlates with earlier infection, and APOE3/4 polymorphisms increase likelihood of severe malaria.[86] Lyme disease Borrelia burgdorferi, the causative agent of Lyme disease, is a host-adapted pathogen that acquires environmental cholesterol to form glycolipids for use in cell membrane maintenance. In one experiment in 2015, mice engineered with apoE deficiency were infected with Borrelia spirochetes. The knockout mice suffered from an increased spirochete burden in joints, as well as inflamed ankles, when compared with wild-type mice. This study suggests that apoE deficiency (and potentially other hyperlipidemias) may be a risk factor in the pathogenicity of Lyme disease. Interactions Interactive pathway map Click on genes, proteins and metabolites below to link to respective articles. [§ 1] 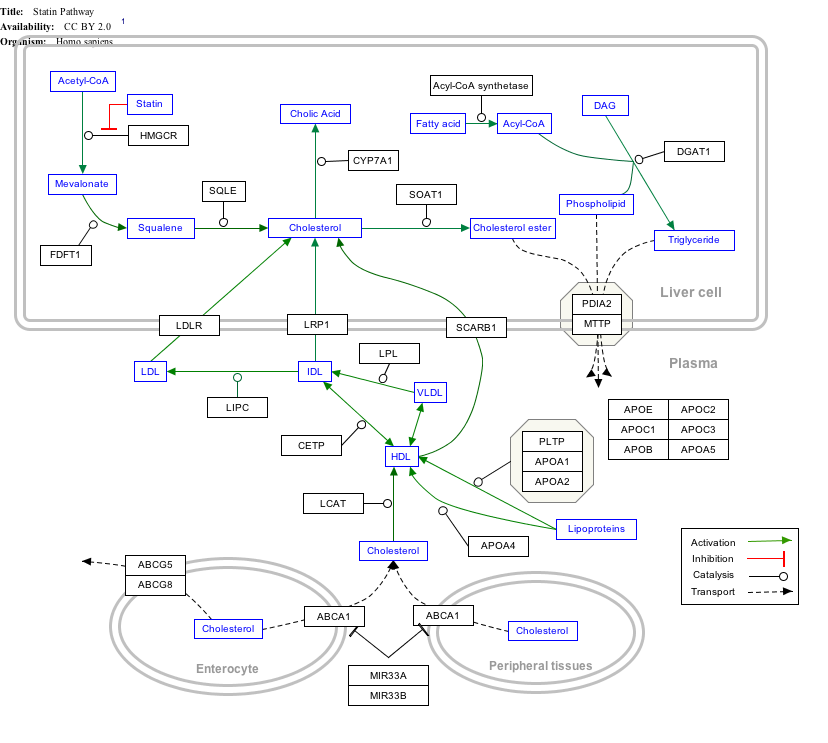 https://en.wikipedia.org/wiki/Apolipoprotein_E |
実
際、アポリポタンパク質ε4アイソフォームは、場合によっては他のアイソフォームよりも認知機能低下に対してより保護的である[58]ので、認知機能、ア
ルツハイマー病の発症、心血管疾患、テロメア短縮などに対するAPOE多型の影響について断定的な発言をする前には注意が必要である。これらの不利な結果
を主張する研究の多くは、再現性のない単独の研究によるものであり、研究はこのアイソフォームに関するチェックされていない仮定に基づいている。2007
年の時点では、APOE多型が若年層の認知に影響を及ぼすという証拠はなく(APOE4が若年層でエピソード記憶能力と神経効率が上昇する可能性があると
いう以外には)、APOE4アイソフォームが感染症のリスクを高めるという証拠もない[59]。 しかしながら、APOE4対立遺伝子とアルツハイマー病との間の関連は、マイノリティグループでは白人グループと比較して弱いことが示されている[9]。 APOE4対立遺伝子のホモ接合体であるヒスパニック系/ラテン系およびアフリカ系アメリカ人は、アルツハイマー病を発症する確率がそれぞれ2.2倍およ び5.7倍であった。 APOE4対立遺伝子は、東アジアの集団ではさらに強い影響を及ぼし、日本人の集団では他の集団と比較して33倍の確率であった[61]。APOE4対立 遺伝子ホモ接合の白人は12.5倍の確率であった[60][9]。 機能 APOEはリポ蛋白脂質輸送システムの構成因子として、血液を介して脂質、脂溶性ビタミン、コレステロールの輸送を促進する。APOEはLDL受容体と相 互作用し、VLDL残渣のエンドサイトーシスを促進する。APOEは主に肝臓で合成されるが、脳、腎臓、脾臓などの他の組織でも見つかっている[21]。 肝臓で合成されたAPOEはHDLと会合し、HDLはAPOEを新しく形成されたVLDLまたはカイロミクロン粒子に分配し、最終的に肝臓への取り込みを 促進する。 神経系では、非神経細胞、特にアストログリアとミクログリアがAPOEの主要な産生細胞であり、一方、神経細胞はAPOEの受容体を優先的に発現する。 APOEは当初、リポ蛋白代謝と心血管疾患における重要性から認識された。APOEの欠損は、カイロミクロン、VLDL、LDLのクリアランス障害の結果 として血漿コレステロールとトリグリセリドが増加する、家族性ジスベタリポ蛋白血症、別名III型高リポ蛋白血症(HLP III)を引き起こす。 [64][7]さらに最近では、アルツハイマー病(AD)、免疫調節、認知など、リポ蛋白輸送とは直接関係のないいくつかの生物学的過程における役割が研 究されている[5]。正確なメカニズムはまだ解明されていないが、APOE対立遺伝子によってコードされるAPOEのアイソフォーム4は、機械的損傷後の カルシウムイオンレベルの上昇とアポトーシスと関連している[65]。 免疫制御の分野では、T細胞増殖の抑制、マクロファージ機能制御、ナチュラルキラーT細胞への(CD1による)脂質抗原提示促進[66]、炎症や酸化の調 節など、多くの免疫学的プロセスにおけるAPOEの相互作用を指摘する研究が増えている。 [67] APOEはマクロファージによって産生され、APOEの分泌はPBMC中の古典的な単球に限定されることが示されており、単球によるAPOEの分泌は炎症 性サイトカインによってダウンレギュレートされ、TGF-βによってアップレギュレートされる[68]。 臨床的意義 アルツハイマー病 2012年の時点で、E4変異体は、様々な民族集団における遅発性散発性アルツハイマー病(AD)の最大の既知の遺伝的危険因子であった。ナイジェリア人 は世界の集団の中でAPOE4対立遺伝子の観察頻度が最も高いが[70]、彼らの間でADが発症することはまれである[70][71]。これはアミロイド との相互作用によって引き起こされる可能性がある[74]。アルツハイマー病は、ペプチドβアミロイドの凝集体の蓄積によって特徴づけられる。アポリポ蛋 白Eは、細胞内と細胞間の両方で、このペプチドの蛋白分解を促進する。APOE-ε4というアイソフォームは、このような反応を促進する効果が他のものほ ど高くないため、この遺伝子変異を持つ個体ではADに対する脆弱性が高くなる[75]。 最近、アルツハイマー病のアミロイド仮説は疑問視されており、Science誌の論文では「煙を除去しても火は消えないように、アミロイド斑を減少させて もアルツハイマー病の経過には影響しないかもしれない」と主張されている。 「E4変異体が担っている役割は、有効なアミロイド仮説がない場合でも、リーリンシグナル伝達がアルツハイマー病に関与する重要なプロセスの1つであるこ とが明らかにされており[77]、E4変異体が神経細胞のリーリン受容体の1つであるApoER2と相互作用してリーリンシグナル伝達を阻害することが示 されていることから、十分に説明することができる[77]。 AD患者の40~65%は少なくとも1コピーのε4対立遺伝子を持っているが、APOE4は疾患の決定因子ではない。AD患者の少なくとも3分の1は APOE4陰性であり、APOE4ホモ接合体の中には発症しない者もいる。しかし、ε4対立遺伝子を2つ持つ患者では、AD発症のリスクが20倍にもなる [78]。また、APOE2対立遺伝子がADにおいて保護的な役割を果たす可能性があるという証拠もある[79]。したがって、アルツハイマー病のリスク が最も高く、かつ早期に発症する遺伝子型はAPOE4,4である。遺伝子型APOE3,3を基準として(この遺伝子型を有する人はリスクレベルが1.0で あるとみなされる)、白人集団のみを対象とした場合、遺伝子型APOE4,4を有する人は、アルツハイマー病を発症するオッズ比が14.9である。 APOE3,4の遺伝子型を持つ人のオッズ比は3.2であり、2対立遺伝子と4対立遺伝子のコピーを持つ人(APOE2,4)のオッズ比は2.6である。 2対立遺伝子と3対立遺伝子をそれぞれ1コピーずつ持つ人(APOE2,3)のオッズ比は0.6である。2対立遺伝子を2コピー持つ者(APOE2,2) もオッズ比0.6である[80]。 ApoE4は、個人がアルツハイマー病を発症する確率を大幅に増加させることが判明しているが、2002年の研究では、APOE対立遺伝子の組み合わせに かかわらず、高血清総コレステロールおよび中年期の高血圧は独立した危険因子であり、これらを合わせると、その個人が後にADを発症する危険性がほぼ3倍 になると結論づけている。 [73] 彼らのデータから予測すると、たとえApoE4対立遺伝子を2つ持っていたとしても、血清コレステロール値を下げることでアルツハイマー病のリスクが低下 する可能性があることを示唆する研究者もいる。 女性は、ほとんどの年齢およびAPOE遺伝子型において、男性よりもADを発症する可能性が高い。ε4対立遺伝子を持つ発症前の女性は、男性よりも有意に神経機能障害が多い[81]。 APOE-ε4は、ADだけでなく、純粋なα-シヌクレイン病変における認知症のリスクも増加させる[82]。 海馬萎縮に対するAPOE-ε4の影響は、より広範な神経変性に先立ち、ADの経過の初期、より軽度の段階でより優勢であることが示唆された[83]。 動脈硬化 アポリポ蛋白E遺伝子を欠損したノックアウトマウス(APOE-/-)は、高脂肪食を与えると極度の高コレステロール血症を発症する[84]。 マラリア APOE-/-ノックアウトマウスは、脳マラリアの顕著な減衰と生存期間の延長を示し、血液脳関門の保護によると思われる脳内の寄生虫とT細胞の隔離の減 少を示す[85]。ヒトの研究では、APOE2多型は早期感染と相関し、APOE3/4多型は重症マラリアの可能性を増加させることが示されている [86]。 ライム病 ライム病の原因菌であるボレリア・ブルグドルフェリは、宿主に適応した病原体であり、環境中のコレステロールを獲得して糖脂質を形成し、細胞膜の維持に使 用する。2015年のある実験では、アポE欠損マウスにボレリア・スピロヘータを感染させた。このノックアウトマウスは、野生型マウスと比較して、関節に おけるスピロヘータの負荷が増加し、足首に炎症が生じた。この研究は、アポE欠損症(および潜在的には他の高脂血症)がライム病の病原性の危険因子である 可能性を示唆している。 相互作用 双方向パスウェイマップ 以下の遺伝子、タンパク質、代謝産物をクリックすると、それぞれの論文にリンクします。[§ 1] |
++
リ ンク
文 献
そ の他の情報
Copyleft, CC, Mitzub'ixi Quq Chi'j, 1996-2099
![]()
☆
 ☆
☆