DNAを種的差異を根拠にする人種主義の誕生
The birth of racism based on DNA as the basis for species differences
DNAを種的差異を根拠にする人種主義の誕生
The birth of racism based on DNA as the basis for species differences
先住民の (政治的に正しい)定義は、選択の問題、つまり最終的にはアイデンティティ(だれもが複 合的アイデンティティを持つ権利)の選択にならざるをえません。で ないと「人種的に」正しいというDNAによるネオ・レイシズムが今後ますます蔓延する可能性がでてくる。ここでのレイシズムとは「人種主義・人種差別主義」と呼ばれているものです。
つまり「DNAからアイデンティ ティが持てる」とい う奇妙な言説を弄する一部の自然人類学者のプロパガンダはますます成功するのではないでしょうか(→疑似科学的レイシズムの復活)。そして今後、文化人類 学者の構築的アイデンティティ論は、ますます分が悪くなるでしょうねぇ。アイデンティティを種的区分にもとづく感情的レイシズムは、LGBTQへの根強い差別のように、克服するのが非常にやっかいになるでしょう。DTの発言を みていたら、かつて公的な場で不可能だった発語が今日可能になっている、つまり、新しい形の人種主義が復活する、おぞましい未来も口を開けている可能性す らあります。
先住の民を祖先に持つことの意味を、無限に薄めれば誰もが先住民と なる(→先住民の不在の証明になる)。先住民が自分たちの定義を自分たちの内部で厳密にすれば、先住民どうしの正当性をもとめて、先住民が先住民であると いう存在証明ができなくなるどころか、先住民であることを認めたくない、非先住民の側が先住民性を否定する根拠に使われてしまう(→先住民を否定するため の正当化の根拠になる)。これらのアポリアの原因のひとつは、先住民が、DNAなどの本質的根拠をもつという信念つまり妄想からきている。それを克服する ためには(→先住民の定義のない2007年の「先住の民の諸権利の国連宣言」UNDRIPを 批准した、つまり、先住民の存在をみとめた、つまり国民国家は、自らの領内に「存在」する先住民への差別是正に努める国際法的責務を担うこととになったた めに)、先住民の定義を、先住の民という空間的特性の条件を最大限にナンセンスなものとして無効化し、先住民の定義を自己アイデンティティに求める必要が でてくる。
他方、先住民であることをアイデン ティティ・ゲームであると主張すると本質主義からはその尊厳を毀損すると非難され構築主義サイド(→文 化決定論)からは理論の弄びだと揶揄される。非先住民であることが先住民であることからどのように批判構成されるのかの議論が共に抜け落ちていな いだろうか?
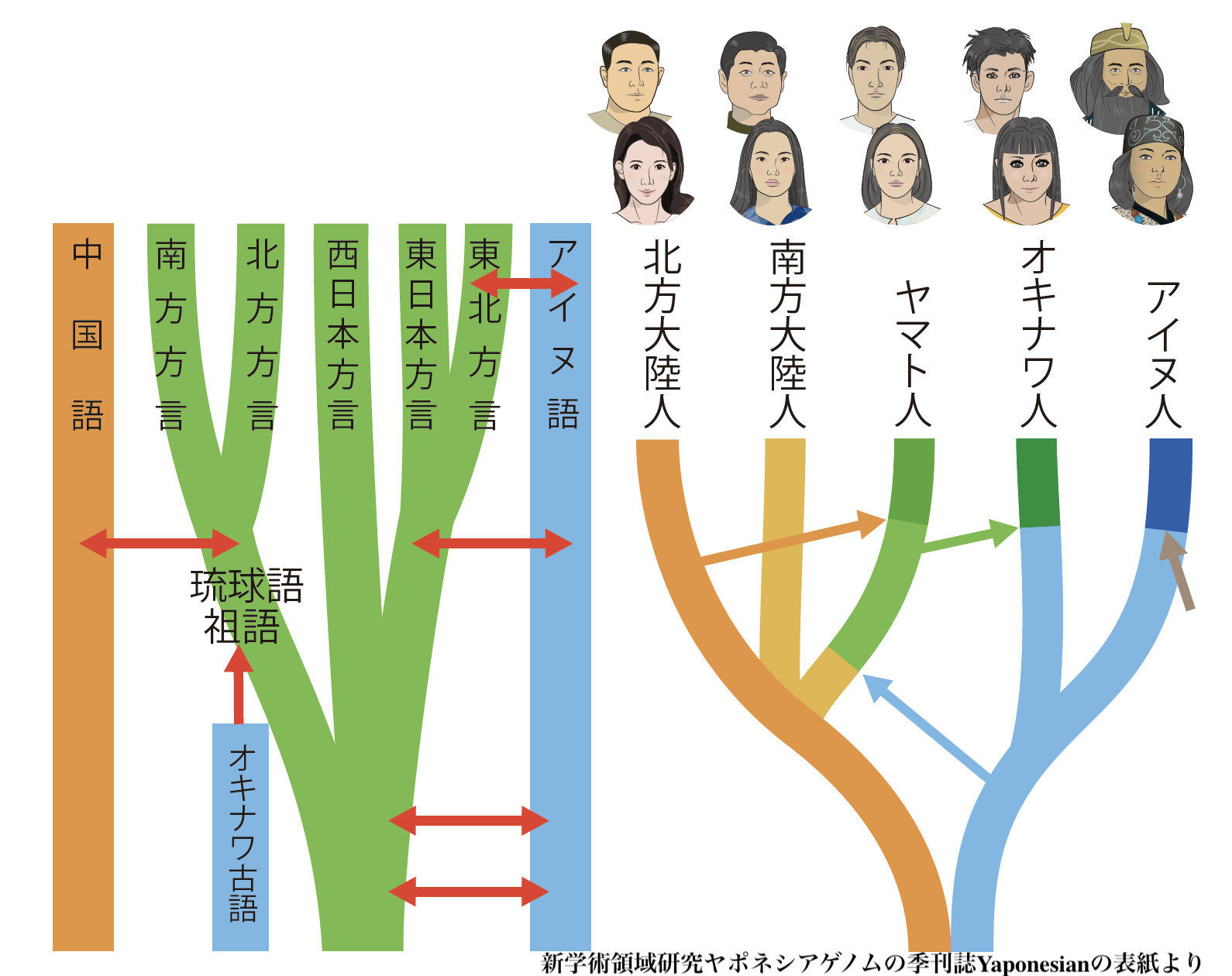
★人間の集団の差をDNAで論証しようとする全地球的プロジェクト(→「ヒトゲノム・プロジェクト」より)
| The 1000
Genomes Project (1KGP),
taken place from January 2008 to 2015, was an international research
effort to establish the most detailed catalogue of human genetic
variation at the time. Scientists planned to sequence the genomes of at
least one thousand anonymous healthy participants from a number of
different ethnic groups within the following three years, using
advancements in newly developed technologies. In 2010, the project
finished its pilot phase, which was described in detail in a
publication in the journal Nature.[1] In 2012, the sequencing of 1092
genomes was announced in a Nature publication.[2] In 2015, two papers
in Nature reported results and the completion of the project and
opportunities for future research.[3][4] Many rare variations, restricted to closely related groups, were identified, and eight structural-variation classes were analyzed.[5] The project united multidisciplinary research teams from institutes around the world, including China, Italy, Japan, Kenya, Nigeria, Peru, the United Kingdom, and the United States contributing to the sequence dataset and to a refined human genome map freely accessible through public databases to the scientific community and the general public alike.[2] The International Genome Sample Resource was created to host and expand on the data set after the project's end.[6] 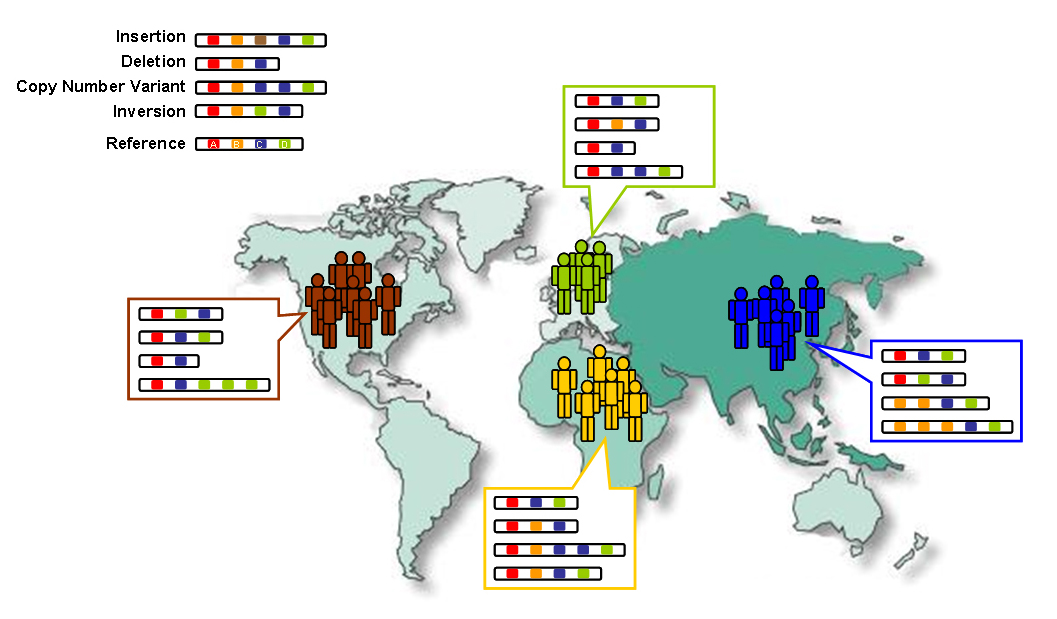 Changes in the number and order of genes (A-D) create genetic diversity within and between populations |
2008
年1月から2015年まで行われた1000人ゲノムプロジェクト(1KGP)は、当時最も詳細なヒトの遺伝的変異のカタログを確立するための国際的な研究
活動であった。科学者たちは、新しく開発された技術の進歩を利用し、その後3年以内に、多くの異なる民族から少なくとも1000人の匿名の健康な参加者の
ゲノムの配列を決定することを計画した。2010年、このプロジェクトは試験段階を終了し、その詳細は学術誌『Nature』に掲載された[1]。
2012年、1092人のゲノムの塩基配列決定が『Nature』誌で発表された[2]。2015年、『Nature』誌に掲載された2つの論文で、プロ
ジェクトの結果と終了、そして今後の研究の可能性が報告された[3][4]。 近縁のグループに限定された多くの稀な変異が同定され、8つの構造変異クラスが解析された[5]。 このプロジェクトは、中国、イタリア、日本、ケニア、ナイジェリア、ペルー、英国、米国など、世界中の研究機関の学際的研究チームを結集し、配列データ セットと、公開データベースを通じて科学界と一般市民が自由にアクセスできる精緻なヒトゲノムマップに貢献した[2]。 国際ゲノム・サンプル・リソース(International Genome Sample Resource)は、プロジェクト終了後にデータセットをホストし、拡張するために設立された[6]。 遺伝子の数と順序の変化(A-D)は、集団内および集団間の遺伝的多様性を生み出す。 |
| Background Since the completion of the Human Genome Project advances in human population genetics and comparative genomics enabled further insight into genetic diversity.[7] The understanding about structural variations (insertions/deletions (indels), copy number variations (CNV), retroelements), single-nucleotide polymorphisms (SNPs), and natural selection were being solidified.[8][9][10][11] The diversity of Human genetic variation such as that Indels were being uncovered and investigating human genomic variations[citation needed] Natural selection It also aimed to provide evidence that can be used to explore the impact of Natural selection on population differences. Patterns of DNA polymorphisms can be used to reliably detect signatures of selection and may help to identify genes that might underlie variation in disease resistance or drug metabolism.[12][13] Such insights could improve understanding of phenotypic variations, genetic disorders and Mendelian inheritance and their effects on survival and/or reproduction of different human populations. Project description This section needs to be updated. Please help update this article to reflect recent events or newly available information. (April 2021) Goals The 1000 Genomes Project was designed to bridge the gap of knowledge between rare genetic variants that have a severe effect predominantly on simple traits (e.g. cystic fibrosis, Huntington disease) and common genetic variants have a mild effect and are implicated in complex traits (e.g. cognition, diabetes, heart disease).[14] The primary goal of this project was to create a complete and detailed catalogue of human genetic variations, which can be used for association studies relating genetic variation to disease. The consortium aimed to discover >95 % of the variants (e.g. SNPs, CNVs, indels) with minor allele frequencies as low as 1% across the genome and 0.1-0.5% in gene regions, as well as to estimate the population frequencies, haplotype backgrounds and linkage disequilibrium patterns of variant alleles.[15] Secondary goals included the support of better SNP and probe selection for genotyping platforms in future studies and the improvement of the human reference sequence. The completed database was expected be a useful tool for studying regions under selection, variation in multiple populations and understanding the underlying processes of mutation and recombination.[15] Outline The human genome consists of approximately 3 billion DNA base pairs and is estimated to carry around 20,000 protein coding genes. In designing the study the consortium needed to address several critical issues regarding the project metrics such as technology challenges, data quality standards and sequence coverage.[15] Over the course of the next three years,[clarification needed] scientists at the Sanger Institute, BGI Shenzhen and the National Human Genome Research Institute’s Large-Scale Sequencing Network planned to sequence a minimum of 1,000 human genomes. Due to the large amount of sequence data that was required, recruiting additional participants was maintained.[14] Almost 10 billion bases were to be sequenced per day over a period of the two year production phase, equating to more than two human genomes every 24 hours. The intended sequence dataset was to comprise 6 trillion DNA bases, 60-fold more sequence data than what has been published in DNA databases at the time.[14] To determine the final design of the full project three pilot studies were to be carried out within the first year of the project. The first pilot intends to genotype 180 people of 3 major geographic groups at low coverage (2×). For the second pilot study, the genomes of two nuclear families (both parents and an adult child) are going to be sequenced with deep coverage (20× per genome). The third pilot study involves sequencing the coding regions (exons) of 1,000 genes in 1,000 people with deep coverage (20×).[14][15] It was estimated that the project would likely cost more than $500 million if standard DNA sequencing technologies were used. Several newer technologies (e.g. Solexa, 454, SOLiD) were to be applied, lowering the expected costs to between $30 million and $50 million. The major support will be provided by the Wellcome Trust Sanger Institute in Hinxton, England; the Beijing Genomics Institute, Shenzhen (BGI Shenzhen), China; and the NHGRI, part of the National Institutes of Health (NIH).[14] In keeping with Fort Lauderdale principles Archived 2013-12-28 at the Wayback Machine, all genome sequence data (including variant calls) is freely available as the project progresses and can be downloaded via ftp from the 1000 genomes project webpage. |
背景 ヒトゲノムプロジェクトが完了して以来、ヒト集団遺伝学 と比較ゲノム学の進歩により、遺伝的多様性についてのさらなる洞察が可能になった[7]。構造変異 (挿入/欠失(インデル)、コピー数変異(CNV)、レトロエレメント)、一塩基多型(SNP)、自然淘汰についての理解が固まりつつあった[8][9] [10][11]。 インデルのようなヒトの遺伝的変異の多様性が明らかにされ、ヒトゲノムの変異が調査されていた[要出典]。 自然淘汰 また、集団の違いに対する自然淘汰の影響を探るために使用できる証拠を提供することも目的としていた。DNA多型のパターンは、淘汰のサインを確実に検出 するために使用することができ、疾患耐性や薬物代謝における変異の根底にある遺伝子を同定するのに役立つ可能性がある[12][13]。このような洞察 は、表現型の変異、遺伝性疾患、メンデル遺伝、およびそれらが異なるヒト集団の生存や繁殖に及ぼす影響についての理解を向上させる可能性がある。 プロジェクトの説明 このセクションは更新が必要である。最近の出来事や新たに入手した情報を反映させるため、この記事の更新にご協力いただきたい。(2021年4月) 目標 1000人ゲノムプロジェクトは、主に単純な形質(嚢胞性線維症、ハンチントン病など)に深刻な影響を及ぼすまれな遺伝的変異と、複雑な形質(認知、糖尿 病、心臓病など)に関与する軽度の影響を及ぼす一般的な遺伝的変異との間の知識のギャップを埋めるために設計された[14]。 このプロジェクトの主な目標は、ヒトの遺伝的変異の完全かつ詳細なカタログを作成し、遺伝的変異と疾患との関連研究に利用できるようにすることであった。 コンソーシアムは、マイナーアレル頻度がゲノム全体で1%、遺伝子領域で0.1-0.5%と低い変異体(SNPs、CNVs、indelなど)の95%以 上を発見し、変異対立遺伝子の集団頻度、ハプロタイプの背景、連鎖不平衡パターンを推定することを目指した[15]。 二次的な目標としては、将来の研究におけるジェノタイピングプラットフォームのためのより良いSNPとプローブの選択のサポート、およびヒトの参照配列の 改良が含まれた[15]。完成したデータベースは、選択下にある領域の研究、複数の集団における変異の研究、変異と組換えの基礎過程の理解に有用なツール となることが期待された[15]。 概要 ヒトゲノムは約30億のDNA塩基対からなり、約20,000のタンパク質コード遺伝子を持つと推定されている。この研究を設計するにあたり、コンソーシ アムは、技術的課題、データ品質基準、配列カバレッジなど、プロジェクトの指標に関するいくつかの重要な問題に取り組む必要があった[15]。 サンガー研究所、BGI深圳、そして国民ゲノム研究所のLarge-Scale Sequencing Networkの科学者たちは、今後3年間で、最低でも1,000のヒトゲノムの配列を決定することを計画した。大量の配列データが必要なため、参加者の 追加募集は維持された[14]。 2年間の製作期間中、1日あたりほぼ100億塩基の塩基配列が決定される予定であり、これは24時間ごとに2人以上のヒトゲノムの塩基配列が決定されるこ とに相当する。意図された塩基配列データセットは、6兆個のDNA塩基で構成される予定であり、当時のDNAデータベースで公開されていた塩基配列データ の60倍であった[14]。 プロジェクト全体の最終的な設計を決定するために、プロジェクトの最初の1年間に3つのパイロット研究が実施されることになっていた。最初のパイロット研 究では、3つの主要な地理的グループの180人を低カバレッジ(2×)で遺伝子型解析する予定である。2つ目のパイロット研究では、2つの核家族(両親と 成人した子供)のゲノムをディープカバレッジ(ゲノムあたり20倍)で配列決定する。3番目のパイロット研究では、1,000人の1,000遺伝子のコー ド領域(エクソン)をディープカバレッジ(20倍)で配列決定する[14][15]。 標準的なDNA配列決定技術を使用した場合、このプロジェクトには5億ドル以上の費用がかかると見積もられている。いくつかの新しい技術(Solexa、 454、SOLiDなど)が適用され、予想されるコストは3,000万ドルから5,000万ドルの間に引き下げられる予定であった。主な支援は、英国ヒン クストンのウェルカム・トラスト・サンガー研究所、中国深センの北京ゲノム研究所(BGI Shenzhen)、および国民衛生研究所(NIH)の一部であるNHGRIが行う予定である[14]。 フォートローダーデールの原則に則り、プロジェクトの進行に伴い、すべてのゲノム配列データ(バリアントコールを含む)が自由に利用できるようになり、 1000ゲノムプロジェクトのウェブページからftp経由でダウンロードできるようになった。 |
Human genome samples Locations of population samples of 1000 Genomes Project.[16] Each circle represents the number of sequences in the final release. Based on the overall goals for the project, the samples will be chosen to provide power in populations where association studies for common diseases are being carried out. Furthermore, the samples do not need to have medical or phenotype information since the proposed catalogue will be a basic resource on human variation.[15] For the pilot studies human genome samples from the HapMap collection will be sequenced. It will be useful to focus on samples that have additional data available (such as ENCODE sequence, genome-wide genotypes, fosmid-end sequence, structural variation assays, and gene expression) to be able to compare the results with those from other projects.[15] Complying with extensive ethical procedures, the 1000 Genomes Project will then use samples from volunteer donors. The following populations will be included in the study: Yoruba in Ibadan (YRI), Nigeria; Japanese in Tokyo (JPT); Chinese in Beijing (CHB); Utah residents with ancestry from northern and western Europe (CEU); Luhya in Webuye, Kenya (LWK); Maasai in Kinyawa, Kenya (MKK); Toscani in Italy (TSI); Peruvians in Lima, Peru (PEL); Gujarati Indians in Houston (GIH); Chinese in metropolitan Denver (CHD); people of Mexican ancestry in Los Angeles (MXL); and people of African ancestry in the southwestern United States (ASW).[14] 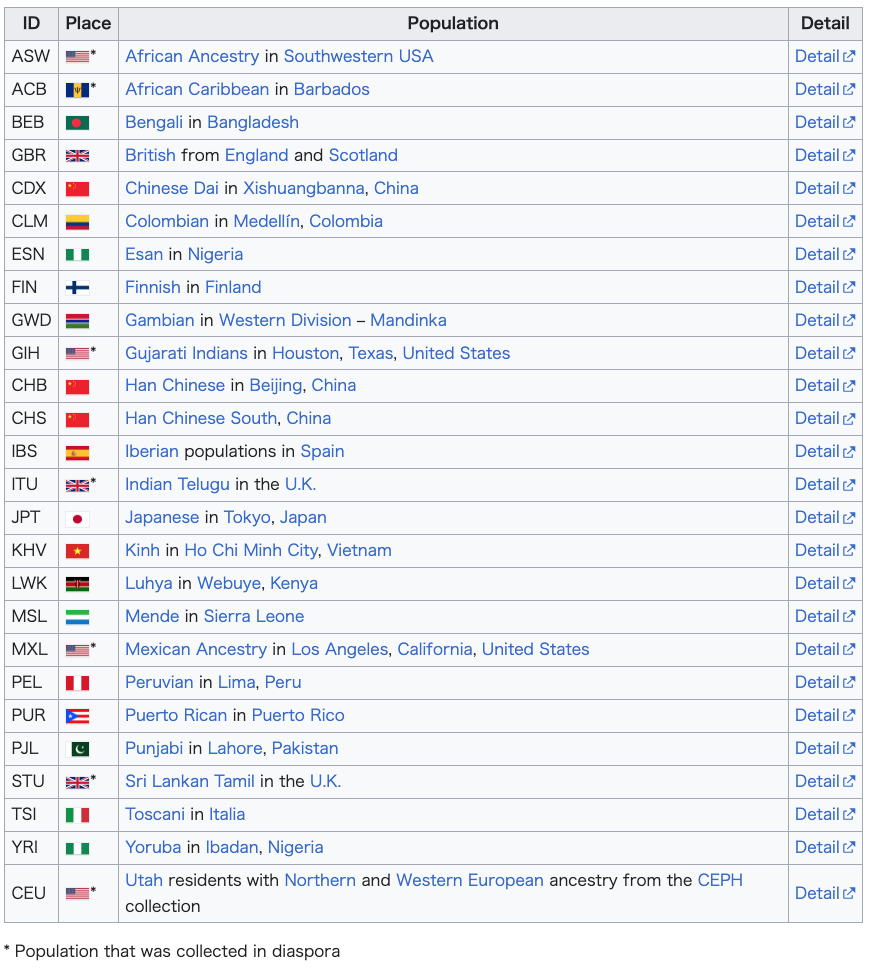 Community meeting Data generated by the 1000 Genomes Project is widely used by the genetics community, making the first 1000 Genomes Project one of the most cited papers in biology.[17] To support this user community, the project held a community analysis meeting in July 2012 that included talks highlighting key project discoveries, their impact on population genetics and human disease studies, and summaries of other large-scale sequencing studies.[18] Project findings Pilot phase The pilot phase consisted of three projects: low-coverage whole-genome sequencing of 179 individuals from 4 populations high-coverage sequencing of 2 trios (mother-father-child) exon-targeted sequencing of 697 individuals from 7 populations It was found that on average, each person carries around 250–300 loss-of-function variants in annotated genes and 50-100 variants previously implicated in inherited disorders. Based on the two trios, it is estimated that the rate of de novo germline mutation is approximately 10−8 per base per generation.[1] |
ヒトゲノムサンプル 1000ゲノムプロジェクトの集団サンプルの位置[16]。各円は最終リリースに含まれる配列数を表す。 プロジェクトの全体的な目標に基づき、サンプルは一般的な疾患の関連研究が行われている集団で力を発揮するように選ばれる。さらに、提案されているカタロ グはヒトの変異に関する基本的なリソースとなるため、サンプルは医学的情報や表現型情報を持っている必要はない[15]。 パイロット研究では、HapMapコレクションからのヒトゲノムサンプルが配列決定される。他のプロジェクトからの結果と比較できるように、追加データ (ENCODE配列、ゲノムワイド遺伝子型、ホスミド末端配列、構造変異アッセイ、遺伝子発現など)が利用可能なサンプルに焦点を当てることは有用であろ う[15]。 広範な倫理的手続きを遵守し、1000人ゲノムプロジェクトはボランティアドナーからのサンプルを使用する。以下の集団が研究の対象となる: ナイジェリアのイバダンに住むヨルバ人(YRI)、東京に住む日本人(JPT)、北京に住む中国人(CHB)、ヨーロッパ北部および西部の祖先を持つユタ 州の住民(CEU)、ケニアのウェブイエに住むルヒヤ人(LWK)、ケニアのキニヤワに住むマサイ人(MKK)、イタリアのトスカーニ人(TSI); ペルーのリマに住むペルー人(PEL)、ヒューストンに住むグジャラティ・インディアン(GIH)、デンバー大都市圏に住む中国人(CHD)、ロサンゼル スに住むメキシコ系祖先を持つ人々(MXL)、アメリカ南西部に住むアフリカ系祖先を持つ人々(ASW)である。 [14] コミュニティミーティング 1000ゲノムプロジェクトによって生成されたデータは、遺伝学のコミュニティで広く利用されており、最初の1000ゲノムプロジェクトは生物学で最も引 用された論文の1つとなっている[17]。このユーザーコミュニティを支援するため、プロジェクトは2012年7月にコミュニティ分析会議を開催し、プロ ジェクトの主要な発見、集団遺伝学やヒト疾患研究への影響、他の大規模シーケンス研究の概要について講演を行った[18]。 プロジェクトの成果 パイロット段階 パイロットフェーズは3つのプロジェクトで構成されていた: 4つの集団から179人の低カバレッジ全ゲノムシーケンス 2つのトリオ(母-父-子)のハイカバレッジシークエンシング 7個体群から697個体のエクソンターゲットシークエンシング その結果、注釈付き遺伝子の機能喪失バリアントは平均して一人当たり約250~300個、遺伝性疾患に以前から関与していたバリアントは50~100個保 有していることが判明した。この2つのトリオに基づくと、de novo生殖細胞突然変異の割合は、1世代あたり1塩基あたり約10-8であると推定される[1]。 |
| Human Genome Project HapMap Project, International HapMap Project Personal genomics Population groups in biomedicine, Race and health 1000 Plant Genomes Project List of biological database |
ヒトゲノムプロジェクト ハップマッププロジェクト 個人ゲノム 生物医学における集団 1000植物ゲノムプロジェクト 生物学データベースのリスト |
| https://en.wikipedia.org/wiki/1000_Genomes_Project |
■fMRIと人種主義(参考)
「「MRI 業界は精力的に利益をあげ、PET
やSPECT
など他のブレイン・イメージング装置とも相まって脳の機能的な構造の解明を驚異的に進めてきた。その成果を利用して、意図、発話、学習などの過程の研究が
始まっている。そして、ついに、fMRIを用いれば、脳の機能と思考や行動との結びつきの体系化が可能だという概念を核として研究産業が展開された[→発
達してきた?引用者]。研究結果の多くは、人間の性質のうちであまりかんばしくはないものと脳内過程との関連を示していて、そういった性質を白日のもとに
曝け出したようにも見える。……たとえば、エリザベス・フェルプス率いるニューヨーク大学の神経科学者、グループは、白人が黒人の顔を見て自動的にネガ
ティブに評価する
ことが、扁桃体の活動と関連していることを示した。刺激を受けると扇桃体が活動して情動を引き起こすのだ。それから、スタンフォード大学の研究者は、顔を
認識する領域の活動性が、自分と同じ人種の顔を見ているときのほうが高いことを発見した。自分と異なる人種に属する典型的な顔を見ると異なるニューロン群
が活性化するという結論が出されたが、シリーズで行われた実験結果はこれを支持している。これらの結果を全部考え合わせてわかってくるのは、次のようなこ
とだろう。「自分たち」と「あの人たち」という概念はかつては適応的だったが、今では対立の種になりかねない、いわば進化の遺物なのだが、いまだに私たち
に組み込まれているのだ。こう考えると、根深い部族主義的な傾向に立ち向かうために断固とした社会政策が必要なのはなぜかわかってくる[→ポストモダンの
ルソー?:引用者]。この他、人間の道徳的決定のしかた、他人に対する共感の程度、自尊心のレベルなどを前頭葉の活動を計測して評価した研究などがある。
中には、民主党員と共和党員がキャンペーンビデオにどのように反応するか調べたものまである」(モレノ 2008:194-195)。」(→「ジョナサン・モレノ『操作される脳』ファンサイト」)
●ナチスはなぜ「人種主義国家」と呼ばれるのか?
その理由は、第三帝国は、体制に帰属できるか否かの
分類に、生物学の「秩序概念」を用いたから。
リンク
文献
その他の情報
Copyleft, CC, Mitzub'ixi Quq Chi'j, 1996-2099
Copyleft, CC, Mitzub'ixi Quq Chi'j, 1996-2099
![]()
☆
 ☆
☆